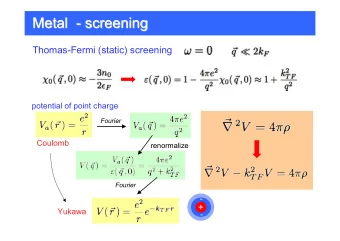
Gaussian ensemble screening (GES): A new Gaussian ensemble screening (GES): A new approach to approach to polypharmacology polypharmacology and virtual and virtual screening screening Violeta I. Pérez-Nueno, Vishwesh Venkatraman, Lazaros Mavridis, David W. Ritchie Orpailleur Team, INRIA Nancy - Grand Est L i : Estrogen L j : Androgen D g i (x) g j (x) σ j σ σ σ σ σ i σ σ CM CM x-x j x-x i x j x i LORIA (Laboratoire Lorrain de Recherche en Informatique et ses Applications), ESCUELA TÉCNICA SUPERIOR INRIA Nancy – Grand Est, 615 rue du Jardin Botanique, 54506 Vandoeuvre-lès-Nancy, France 1/31 1 /31
Polypharmacology Polypharmacology (Drug selectivity) Multiple drugs bind to a given target A given drug binds to more than (promiscuous targets) one target (promiscuous ligands) Promiscuous Ligand Promiscuous Ligand Promiscuous Target 2/31 2 /31
Previous work Relate receptors to each other quantitatively based on the similarity in the: Sequence space Ligand space Binding pocket space (chemical fingerprints) (pharmacophoric descriptors) •Keiser et al . Nature Biotechnol. 2007 , 25 , 197-206. Similarity Ensemble Approach (SEA) relates proteins based on the set-wise chemical similarity among their ligands. •Vidal & Mestres. Mol. Inf. 2010 , 29, 543. PHRAG, FPD, SHED molecular descriptors. •Weskamp et al. Proteins 2009 , 76 , 317-330. Similarity amongst binding pockets extracted by LIGSITE algorithm. •Milletti, F.; Vulpetti, A. J. Chem. Inf. Model., 2010 , 50 , 1418–143. Binding pocket comparison using four-point pharmacophoric descriptors based on GRID. 3/31 3 /31
Our approach Gaussian Ensemble Screening (GES): 3D spherical harmonic (SH) shape-based approach which compares molecular surfaces and predicts quantitatively the relationships between drug classes very fast and efficiently. ! !"# $% & ' ( & )& *+, % - . /% & ' ( & ) & *+, % !"# $% & ' ( & )& *+, % ! ! - . /% - . /% & & ' ( & ' ( & ) & ) & *+, % *+, % ! ! ! ! ! ! ! 0123 # 4% 2' *56+' & 7 *% "89: +*, & 8' 8% ; <' *( 87 & 7% !' (& )& *+, % 0123 # 4% 2' *56+' & 7 *% ! "89: +*, & 8' 8% ; <' *( 87 & 7% !' (& )& *+, % ! ! ! ! ! ! ! 15, )5= 8>( 8? % 18>( 5@ +7 >+, & ' % 15, )5= 8>( 8? % ! 18>(5@ +7 >+, & ' % ! ! ! ! ! ! B ! ! % B ! ! ! % ! 2' *& 5' A, +68' % ; *8, +& A% ! ! , 8A9=*57 8% ; *8, +& A% , 8A9= *57 8% 2' *& 5' A, +68' % ! 4/31 4 /31
Methodology 1. Calculating SH consensus shapes and center molecules 2. Ligand set representations 3. Gaussian ligand set comparisons C C M M 4. Finding the best clustering threshold 5. Gaussian p-values p-value s 6. MDDR polypharmacology interaction matrix 7. Examples of strongly related targets 5/31 5 /31
1. Calculating spherical harmonic shapes Surface shapes are represented as radial distance expansions of the molecular surface with respect to the center of the molecule. • Real SHs: • Coefficients: • Encode radial distances from origin as SH series… • • Solve coefficients by Solve coefficients by numerical integration… Ritchie, D.W. and Kemp, G.J.L. J. Comp. Chem. 1999 , 20, 383–395. 6/31 6 /31
2. Calculating SH consensus shapes and center molecules ) = 1 N L l k y lm θ , ϕ ∑ ∑ ∑ ( ( ) r θ , ϕ a lm N k = 1 l = 0 m =− l “Consensus” shape Pérez-Nueno et al . J. Chem. Inf. Model. 2008 , 48 , 2146–2165. 7/31 7 /31
3. Ligand set representations The idea is to represent a cluster of molecules as a Gaussian distribution with respect to a selected centre molecule (CM). - Calculate SH molecular surfaces of each ligand in each ligand set and superpose them. - Calculate the center molecule (CM) of the ligand set and the normalised SH distance (1-Similarity Score) between that of the CM and each cluster member. - Assuming that these distances follow a Gaussian distribution, each cluster may be represented as a probability density function g i (x) L i : Estrogen 2 ( ) g i (x) x − x i 1 ( ) = 2 g i x 2 ⋅ e 2 σ i 2 πσ i CM σ σ σ σ i σ : SD of the member distances x-x i x i An illustration of a Gaussian ligand set cluster. 8/31 8 /31
4. Gaussian ligand set comparisons By considering the SD of the member distances as the Gaussian width of a distribution, we calculate a “distance” ( D ) between two clusters, i and j , and normalizing the distance term we can write it as a Hodgkin-like similarity score Sij between two distributions. 12 +∞ − a ⋅ b 32 ⋅ a ⋅ b 2 ∫ ⋅ x ij ( ) ⋅ g j x ( ) dx g i x 2 2 ⋅ e a + b a + b a=1/2 σ i 2 S ij = −∞ S ij = ( ) b=1/2 σ j 2 2 dx + 2 dx +∞ +∞ 12 + b 12 ∫ ∫ ( ) ( ) a g i x g j x x ij : distance between the CMs of clusters i and j −∞ −∞ L i : Estrogen L j : Thrombin D D g i (x) g j (x) σ σ σ j σ σ σ i σ σ CM CM x-x j x i x j x-x i S ij = 1.33 ! 10 ″ 41 Illustration of the very small Gaussian overlap between the estrogen and thrombin ligand sets. 9/31 9 /31
4. Gaussian ligand set comparisons The similarity between drug classes can be calculated rapidly and reliably by calculating the Gaussian overlap between pairs of such clusters. Thus, it is straight-forward to calculate all-against-all cluster comparisons. It is worth noting that our cluster similarity score depends only on the similarity of pairs of centre molecules and the SDs of their respective clusters. It does not depend on the number of members of each cluster . L i : Estrogen L j : Androgen D g i (x) g j (x) j σ j σ σ σ σ i σ σ σ CM CM x j x i x-x j x-x i S ij = 0.57 Illustration of the large Gaussian overlap between the estrogen and androgen ligand sets. 10/31 10 10 10 /31
4. Gaussian ligand set comparisons 1. MDDR ANNOTATION FAMILIES SPACE THERAPEUTIC ANNOTATION L 5 : δ δ δ opioid agonist δ L 7 : Dopamine L 1 : Thrombin L 270 : Histamine D3 antagonist H3 antagonist L 2 : Estrogen . . . L 6 : 5HT2A L : 5HT2A L 4 : Gaba α α subunit α α L 8 : Cytocrome P450 antagonist L 8 : Muscarinic Oxidase Inhibitor L 3 : Androgen M2 antagonist We applied the approach to 270 specific therapeutic annotations in MDDR. Ligands which share an annotation define a set of functionally related molecules which we call a “ligand set”. MDDR annotations are quite general and were primarily derived from the patent literature. A given annotation may thus contain a diverse set of compounds with a wide range of affinities. 11/31 11 11 11 /31
4. Gaussian ligand set comparisons 2. MDDR ANNOTATION SHAPE CLUSTERS L 1 C3 L 7 C1 L 1 C2 L 5 C1 L 267 C1 L 1 C1 L 267 C2 CM C3 L 5 C2 CM C2 L 7 C2 L 2 C1 . . . C M C L 8 C1 1 L 6 C1 L 4 C1 L 6 C2 L 3 C1 L 8 C1 L 8 C2 L 8 C3 L 6 C3 L 6 C4 L 8 C4 L 8 C5 L 8 C6 ANNOTATION CLUSTER In order to eliminate outliers, we used the CAST clustering algorithm to cluster the members of each annotation using their PARAFIT Tanimoto similarity scores. We then calculated the consensus shape and the center molecule for each cluster, and we eliminated any cluster members beyond 1.5 standard deviations (SDs) from the corresponding CM. 12/31 12 12 12 /31
5. Finding the best clustering threshold 2. MDDR ANNOTATION SHAPE CLUSTERS L 1 C3 L 7 C1 L 1 C2 L 5 C1 L 267 C1 L 1 C1 L 267 C2 CM C3 B CM C2 B L 5 C2 CM C3 A L 7 C2 CM C2 A L 2 C1 . . . CM C1 A CM C1 B L 1 C1 B L C1 L C1 L 1 C1 A L 8 C1 L 6 C1 L 4 C1 L 6 C2 L 3 C1 L 8 C1 L 8 C2 L 8 C3 L 6 C3 L 6 C4 L 8 C4 L 8 C5 L 8 C6 ANNOTATION CLUSTER We clustered each annotation according to Parafit Shape Tanimoto using different similarity thresholds: 0.6, 0.65, 0.675, 0.7, 0.8, 0.85. Each ligand set was randomly split into two almost equally sub-clusters, and all- vs- all clustering was performed with the aim of split and reassemble the split clusters correctly. 13/31 13 13 13 /31
5. Finding the best clustering threshold L 2 C1 (Estrogen) L 1 C1 (Thrombin) L 3 C1 (Androgen) L 2 C1 A L 2 C1 B L 1 C1 A L 1 C1 B L 3 C1 A L 3 C1 B CM C1 B CM C1 A CM C1 B CM C1 A CM C1 A CM C1 B x-x j x-x i X-X i x-x j x-x j x-x i L 4 C1 (Gaba a subunit) L 4 C1 A L 4 C1 B 3. SPLIT ANNOTATION SHAPE CLUSTERS + GAUSSIAN SCORING . . . I.e. here are shown the C1 of different annotations split in two x-x j groups to obtain the distribution of scores for the true cases, CM C1 CM C1 B where annotations are related to each other (L 1 C1 A vs L 1 C1 B , A L 2 C1 A vs L 2 C1 B ...) , and the false cases, where the annotations x-x i are not related (L 1 C1 A vs L 2 C1 A , L 1 C1 A vs L 3 C1 A ...). If we can split and reassemble clusters of molecules that we know they are related, then we can identify interesting relationships between clusters of molecules that we don’t know they are related 14/31 14 14 14 /31
Recommend
More recommend
Unleash a World of Digital Possibilities—Browse, Share, and Explore Content Without Boundaries