
Protein Folding Simulation in Concurrent Constraint Programming - PowerPoint PPT Presentation
Protein Folding Simulation in Concurrent Constraint Programming Luca Bortolussi, Alessandro Dal Pal` u, Agostino Dovier DIMI, Univ. of Udine (IT) Federico Fogolari DST, Univ. of Verona (IT) Outline of the talk Introduction Concurrent
Protein Folding Simulation in Concurrent Constraint Programming Luca Bortolussi, Alessandro Dal Pal` u, Agostino Dovier DIMI, Univ. of Udine (IT) Federico Fogolari DST, Univ. of Verona (IT)
Outline of the talk • Introduction • Concurrent framework • Testing model • Results • Future Work L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 2/20
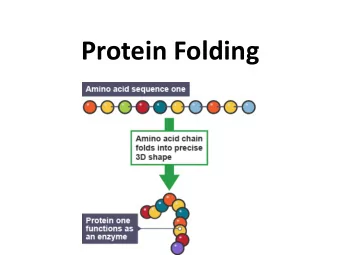
Proteins Proteins are abundant in nature and fundamental to life. • • The diversity of 3D protein structure underlies the very large range of their function (Enzymes, Storage, Transport, Mes- sengers, Antibodies, Regulation, mechanical support). • A Protein is a polymer chain made of monomers ( aminoacids ) of 20 different kinds. Aminoacids have a common part (6 atoms) and a distinguish- • ing part (from 1 to 18 atoms). • They are typically identified by one letter in { A, . . . , Z }\{ B, J, O, U, X, Z } . L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 3/20
Proteins The Primary Structure is the sequence of aminoacids consti- • tuting a protein. • The Secondary Structures of a Protein are local structures ( α - helices , β -sheets ) which formation is caused by local forces. • The Tertiary Structure , that determines macroscopic proper- ties and biological functions, is the 3D conformation of the Protein. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 4/20
Example: Protein 1ENH Primary Structure: • R,P,R,T,A,F,S,S,E,Q, L,A,R,L,K,R,E,F,N,E, N,R,Y,L,T,E,R,R,R,Q, Q,L,S,S,E,L,G,L,N,E, A,Q,I,K,I,W,F,Q,N,K, R,A,K,I • Tertiary Structure: All atom Model / Simplified Model L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 5/20
The Protein Structure Prediction Problem • Proteins fold in a determined environment (e.g. water) to form a very specific geometric pattern ( native state/conformation ). • The native conformation is relatively stable and unique, and corresponds to a state which minimizes the global free energy. • The Protein Structure Prediction problem (PSP) consists in pre- dicting the Tertiary Structure of a protein, given its Primary Structure. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 6/20
Approaches to PSP Several different approaches have been used to tackle PSP: • Homology modelling and folding recognition; • All atoms simulation using molecular dynamics; • Constraint-based approaches in lattices; • Ab-initio simulations using simplified models. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 7/20
CCP simulation framework • We encoded the PSP problem into a Concurrent Constraint (Logic) Programming paradigm. • Each aminoacid is associated to an independent process. • Each process communicates with the others, and reacts to their changes of the spatial position. • The framework is independent from the spatial model of the protein and from the energy model. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 8/20
Communication Strategy • Each process, before performing a move, waits for the commu- nication of a movement of some other aminoacid; • The information of position changes of process P i is stored in a list L i of logic terms (leaving the tail variable uninstantiated), thus keeping track of the entire history of the folding known to him; • Each process, while moving, uses the most recent information available to him, i.e. the last ground terms of the lists L i ; • each process, once it has moved, communicates to all other processes its new position updating its list. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 9/20
Abstract CCP program 7 run(ID, S, [P1, P2, ..., Pn]):- 1 simulation(S):- 8 getTails([P1, ..., Pn],[T1, ..., Tn]), 2 Init=[[I1|_], 9 ask(T1=[_|_]) -> skip + [I2|_], 10 ask(T2=[_|_]) -> skip + ..., 11 ... + [In|_]], 12 ask(Tn=[_|_]) -> skip, 3 run(1,S,Init) || 13 getLast([P1, ..., Pn],[L1, ..., Ln]), 4 run(2,S,Init) || 14 updatePosition(ID,S,[L1,..,Ln],NP), 5 ... || 15 tell(TID=[NP|_]), 6 run(n,S,Init). 16 run(ID,S,[P1, ..., Pn]). • The main clause is simulation . • Init is a variable containing n lists which contain the initial positions Ii . • n concurrent calls to run are called, one for each process - aminoacid. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 10/20
Abstract CCP program 7 run(ID, S, [P1, P2, ..., Pn]):- 1 simulation(S):- 8 getTails([P1, ..., Pn],[T1, ..., Tn]), 2 Init=[[I1|_], 9 ask(T1=[_|_]) -> skip + [I2|_], 10 ask(T2=[_|_]) -> skip + ..., 11 ... + [In|_]], 12 ask(Tn=[_|_]) -> skip, 3 run(1,S,Init) || 13 getLast([P1, ..., Pn],[L1, ..., Ln]), 4 run(2,S,Init) || 14 updatePosition(ID,S,[L1,..,Ln],NP), 5 ... || 15 tell(TID=[NP|_]), 6 run(n,S,Init). 16 run(ID,S,[P1, ..., Pn]). • ID is the identification code of the aminoacid. • getTails gets the tails of the lists P1,...,Pn and assigns them to the variables T1,...,Tn . • Then the process waits for one of these variables to be instan- tiated with ask(Ti=[_|_]) -> skip . L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 11/20
Abstract CCP program 7 run(ID, S, [P1, P2, ..., Pn]):- 1 simulation(S):- 8 getTails([P1, ..., Pn],[T1, ..., Tn]), 2 Init=[[I1|_], 9 ask(T1=[_|_]) -> skip + [I2|_], 10 ask(T2=[_|_]) -> skip + ..., 11 ... + [In|_]], 12 ask(Tn=[_|_]) -> skip, 3 run(1,S,Init) || 13 getLast([P1, ..., Pn],[L1, ..., Ln]), 4 run(2,S,Init) || 14 updatePosition(ID,S,[L1,..,Ln],NP), 5 ... || 15 tell(TID=[NP|_]), 6 run(n,S,Init). 16 run(ID,S,[P1, ..., Pn]). • Once this happens it retrieves the last information with getLast . • Then it updates its position with updatePosition and communi- cates its move to all other processes by means of tell . • Finally the run procedure is called recursively. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 12/20
Movement Strategy The procedure updatePosition works in the following way: • The aminoacid randomly chooses a new position, close to the current one within a given step; • Using the most recent information available about the spatial position of other processes, it computes the energy relative to the choice; • It accepts the position using a Montecarlo criterion: - If the new energy is lower than the current one, it accepts the move; - If the new energy is greater than the current one, it accepts the move with probability e − Enew − Ecurrent . T • This procedure depends on the spatial model adopted. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 13/20
Movement Strategy The new position is randomly selected in the following way: • We calculate the set of points which keep fixed the distance with the adjacent neighbours of the aminoacid (a circumference or a sphere); • We randomly select a point in this set, close to the current position; • We randomly select a small offset from this point. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 14/20
Testing Model ✬ ✩ side chain ⑦ Cα ❳ C ′ ✘ ❳❳ ❳❳ ✿ ✘ ✘ N ✘✘ ✘ ③ ❳ H ✫ ✪ H O • Each aminoacid is represented as a single center of interaction, which corresponds to the C α atom. • The energy function consists of four terms, which take into account local and global interactions. • This model is very simple, but served as a test for the framework. L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 15/20
Energy Function The Energy Function we use is E ( � s ) = η b E b ( � s ) + η a E a ( � s ) + η t E t ( � s ) + η c E c ( � s ) E b ( � s ) is the Bond Distance term � 2 � � E b ( � s ) = r ( s i , s i +1 ) − r 0 1 ≤ i ≤ n − 1 E a ( � s ) is the Bond Angle Bend term 1 0.8 � 2 � 2 n − 2 � βi − β 1 � βi − β 2 0.6 − − � σ 1 σ 2 0.4 E a ( � s ) = − log + a 2 e a 1 e 0.2 i =1 0 0 1 2 3 Radians L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 16/20
Energy Function The Energy Function we use is E ( � s ) = η b E b ( � s ) + η a E a ( � s ) + η t E t ( � s ) + η c E c ( � s ) E t ( � s ) is the Torsional Angle term (Φ i − φ 1)2 (Φ i − φ 2)2 n − 3 ( σ 1+ σ 0)2 + a 2 e ( σ 2+ σ 0)2 � E t ( � s ) = − log a 1 e i =1 E c ( � s ) is the Contact Interaction term � 12 � 6 n − 3 n � r 0 ( s i , s j ) � r 0 ( s i , s j ) � � E c ( � s ) = | Pot ( s i , s j ) | + Pot ( s i , s j ) r ( s i , s j ) r ( s i , s j ) i =1 j = i +3 1 0.8 Potential 0.6 0 0.4 0.2 0 0 r 1r 2 r 3 r 0 2 4 6 Radians L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 17/20
Implementation The code is implemented in Mozart. There are two classes: • Protein , implements the simulation predicate and coordinates the process associated with the single aminoacids; • Amino , which describes the single aminoacid, and implements all the methods related to the action, like updatePosition , ask and tell . L. Bortolussi, A. Dal Pal` u, A. Dovier, and F. Fogolari BIOCONCUR 2004 — 18/20
Recommend





















More recommend
Explore More Topics
Stay informed with curated content and fresh updates.