
Protein design Chris Bystroff Biology 12 Apr 2016 1 Protein - PowerPoint PPT Presentation
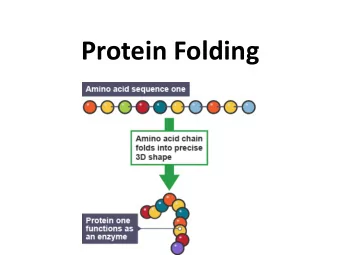
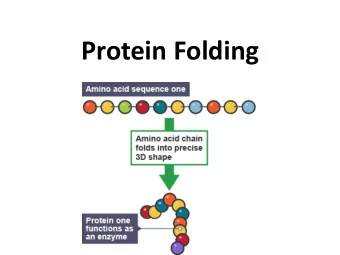
Protein design Chris Bystroff Biology 12 Apr 2016 1 Protein folding/ protein design folding design structure sequence Sequence space maps to structure space sequence families fold space Structure prediction is "many-to-one".
Protein design Chris Bystroff Biology 12 Apr 2016 1
Protein folding/ protein design folding design structure sequence
Sequence space maps to structure space sequence families fold space Structure prediction is "many-to-one".
Sequence space maps to structure space sequence families fold space Design is "one-to-many". Much easier
rational design Binary patterning in proteins (Kamtekar et al, Science, 1993)
P/NP Kamtekar, Satwik, et al. "Protein design by binary patterning of polar and nonpolar amino acids." Science 262.5140 (1993): 1680-1685.
Rationally designed proteins are usually unstable folded energy unfolded folding
Computationally designed proteins are often very stable Circular dichroism spectra measure stability unfolded folded Q:Why? Dantas et al., J. Mol. Biol. (2003) 332, 449–460 A: Better packing.
Computational protein design using Dead-End Elimination naive design algorithm 1. Select positions for mutating. 2. Select allowed amino acids at those positions. 3. For the selected amino acids, try all sidechain orientations ( rotamers) . 4. Chose the sequence of amino acids that gives the lowest energy.
REMINDER: Sidechain Rotamers Sidechain conformations fall into three classes called rotational isomers, or rotamers . A random sampling of Phenylalanine sidechains, w/ backbone superimposed
Sidechain rotamers 1-4 interactions differ greatly in energy depending on the moieties involved. N N N G C CG H CA H H CA CA H CB CB CB H H O = C O = C H O = C H H C G "m" "p" "t" -60° gauche 180° anti/trans +60° gauche
Rotamer stability is dependent on the backbone φψ angles W sidechain is shown here lying over Thr backbone Rotamers of W*: φ ψ P| φ =-140, ψ =160 P| φ =-60, ψ =-40 p-90 +60 -90 0.372 0.079 p90 +60 +90 0.238 0.005 t-105 180 -105 0.033 0.251 t90 180 90 0.021 0.268 m0 -65 5 0.038 0.124 m95 -65 95 0.183 0.203
Rotamer Libraries Rotamer libraries have been compiled by clustering the sidechains of each amino acid over the whole database. Each cluster is a representative conformation (or rotamer), and is represented in the library by the best sidechain angles ( chi angles), the "centroid" angles, for that cluster. Two commonly used rotamer libraries : *Jane & David Richardson: http://kinemage.biochem.duke.edu/databases/ rotamer.php Roland Dunbrack: http://dunbrack.fccc.edu/bbdep/index.php
sidechain prediction using a rotamer library Given the sequence and only the backbone atom coordinates, accurately model the positions of the sidechains . fine lines = true structure thick lines = sidechain predictions Desmet et al, Nature v.356, pp339-342 (1992)
Theoretical complexity of sequence design Total number of sidechain rotamers: R=193 Typical small protein length: L=100 residues Sequence complexity: 20 100 = 1.3*10 130 Rotamer complexity: 193 100 = 3.6*10 228 Complexity of DEE algorithm: O( R 2 L 2 ) = 3.6*10 8
Energy is pairwise j i E ij = Σ (H-bonds+collisions+electrostatics+SAS) SAS = solvent accessible surface 16
Dead end elimination theorem •Each residue is numbered ( i or j ) and each residue has a set of rotamers ( r, s or t ). So, the notation i r means "choose rotamer r for position i ". •The total energy is the sum of the three components: fixed-movable fixed-fixed movable-movable E global = E template + Σ i E( i r ) + Σ i Σ j E( i r ,j s ) where r and s are any choice of rotamers. NOTE: E global ≥ E GMEC for any choice of rotamers.
Dead end elimination theorem •If i g is in the GMEC and i t is not, then we can separate the terms that contain i g or i t and re-write the inequality. E GMEC = E template + E( i g ) + Σ j E( i g ,j g ) + Σ j E( j g ) + Σ j Σ k E( j g ,k g ) ...is less than... E notGMEC = E template + E( i t ) + Σ j E( i t ,j g ) + Σ j E( j g ) + Σ j Σ k E( j g ,k g ) Canceling all terms in black, we get: E( i r ) + Σ j E( i r j s ) > E( i g ) + Σ j E( i g ,j s ) So, if we find two rotamers i r and i t , and: E( i r ) + Σ j min s E( i r j s ) > E( i t ) + Σ j max s E( i t ,j s ) Then i r cannot possibly be in the GMEC.
Dead end elimination theorem E( i r ) + Σ j min s E( i r j s ) > E( i t ) + Σ j max s E( i t ,j s ) DEE theorem can be translated into plain English as follows: If the "worst case scenario" for t is better than the "best case scenario" for r , then you always choose t .
DEE algorithm E global = E template + Σ i E( i r ) + Σ i Σ j E( i r ,j s ) r 1 1 2 3 a b c a b c a b c E(r 2 ) -1 1 1 -2 2 5 a 0 1 b 3 5 1 0 5 -1 0 x c 5 5 -1 0 0 0 5 x 3 x -1 3 5 0 0 1 0 a x x x 1 5 5 12 5 0 r 2 b E(r 1 ,r 2 ) 0 2 a 1 c 1 1 -1 4 3 0 0 b 0 c -2 0 0 0 12 4 a 0 b 2 5 0 0 5 3 3 2 c 5 -1 0 1 0 0 12 0 0 5 0 0 0 0 0 12 E(r 1 ) Find two columns (rotamers) within the same residue, where one is always better than the other. Eliminate the rotamer that can always be beat. (repeat until only 1 rotamer per residue)
r 1 1 2 3 a b c a b c a b c E(r 2 ) -1 1 1 -2 2 5 a 0 1 b 3 5 1 0 5 -1 0 c 5 5 -1 0 0 0 5 -1 3 5 0 0 1 0 a r 2 1 5 5 12 5 0 b E(r 1 ,r 2 ) 2 0 c 1 1 -1 4 3 0 0 0 -2 0 0 0 12 4 a b 2 5 0 0 5 3 0 3 c 5 -1 0 1 0 0 12 0 0 5 0 0 0 0 0 12 E(r 1 )
DEE algorithm E global = E template + Σ i E( i r ) + Σ i Σ j E( i r ,j s ) r 1 1 2 3 a b c a b c a b c E(r 2 ) -1 1 1 -2 2 5 a 0 1 b 3 5 1 0 5 -1 0 c 5 5 -1 0 0 0 5 3 -1 3 5 0 0 1 0 a 1 5 5 12 5 0 r 2 b E(r 1 ,r 2 ) 0 2 a 1 c 1 1 -1 4 3 0 0 b 0 c -2 0 0 0 12 4 a 0 b 2 5 0 0 5 3 3 2 c 5 -1 0 1 0 0 12 0 0 5 0 0 0 0 0 12 E(r 1 ) Find two columns (rotamers) within the same residue, where one is always better than the other. Eliminate the rotamer that can always be beat. (repeat until only 1 rotamer per residue)
Sequence design using DEE r 1 3 1 2 a b a c b a c b a b -1 1 1 -2 1 5 2 0 a 3 5 1 0 5 -1 2 0 b 1 E(r 2 ) 5 5 -1 0 0 0 3 c 5 -1 3 5 0 0 1 1 0 a 1 5 5 12 5 0 -3 0 E(r 1 ,r 2 ) r 2 b 2 1 1 -1 4 3 0 1 0 3 c 0 -2 0 0 0 12 4 a a 0 1 Asp 2 5 0 0 5 3 b b 12 3 a 5 -1 0 1 0 0 c 2 b 2 2 3 1 -3 1 Leu 2 0 0 5 0 0 0 0 0 12 2 E(r 1 ) “Rotamers” within the DEE framework can have different atoms. i.e. they can be different amino acids . Using DEE, we choose the best set of rotamers. Now we have the sequence of the lowest energy structure. In the example, we have D or L at position 3.
DEE with alternative sequences and ligands Ligand conformers . r 1 3 1 2 a b c a b c a b a b -1 1 1 -2 1 5 2 0 a 3 5 1 0 5 -1 2 0 b L E(r 2 ) 5 5 -1 0 0 0 3 c 5 -1 3 5 0 0 1 1 0 a 1 5 5 12 5 0 -3 0 E(r 1 ,r 2 ) r 2 b 2 1 1 -1 4 3 0 1 0 L 3 c 0 -2 0 0 0 12 4 a a 0 Asp 2 5 0 0 5 3 b b 12 3 a 5 -1 0 1 0 0 c 2 b 2 2 3 1 -3 1 Leu 2 0 0 5 0 0 0 0 0 12 2 E(r 1 ) Each alternative ligand position is another “rotamer”.
Case study: designing a seratonin sensor. 1. find a template The native ligand (arabinose) is approximately the same size as the targeted ligand (seratonin). Looger, L. L., Dwyer, M. A., Smith, J. J. & Hellinga, H. W. Nature 423, 185–190 (2003).
2. carve out space for the ligand All sidechains in the binding site were truncated to alanines, and a space was defined (yellow) for the new ligand. Lots of possible ligand orientations were made. Ligand orientations were treated like rotamers in DEE! Looger, L. L., Dwyer, M. A., Smith, J. J. & Hellinga, H. W. Nature 423, 185–190 (2003).
3. Select/place side chains using DEE The most critical component of the energy function was hydrogen bonding (dotted lines). Every donor/acceptor should be satisfied. Looger, L. L., Dwyer, M. A., Smith, J. J. & Hellinga, H. W. Nature 423, 185–190 (2003).
Getting the H-bonding right is key Note plenty of backbone-ligand H-bonds in these successful designs.
New directions for protein design • Docking and design at the same time! • Designing enzymes! • Designing nano-structures!! • Designing drugs (biologics)!!!
My research: Leave-one-out biosensors A B C
Green Fluorescent Protein 11-standed beta-barrel surrounding fluorescent chromophore
GFP biosensor showing bound target pepide
Leave-one-out GFP "LOO-GFP" Change the sequence of one strand to target sequence Design around it Make it in the lab. Test for binding. in vivo expression/co-expression of GFP constructs permuted s7 LOO11 LOO7 LOO11 control control permuted LOO7+ LOO11 LOO7+ LOO11 s7 s11 control control s7 +s11 s11 +s7 LOO7-GFP is circularly permuted GFP with a C-N linker peptide (red arrow), and β -strand 7 is removed (black arrow).
Recommend






















More recommend
Explore More Topics
Stay informed with curated content and fresh updates.