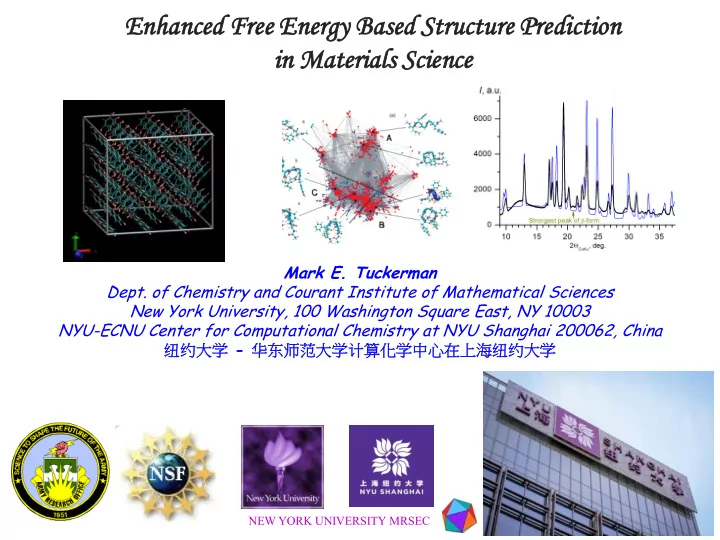
in Material erials s Science ce Mark E. Tuckerman Dept. of - PowerPoint PPT Presentation
Enhanced En nced Free En Energy Based Structure ture Predicti iction on in Material erials s Science ce Mark E. Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Sciences New York University, 100 Washington Square East,
Enhanced En nced Free En Energy Based Structure ture Predicti iction on in Material erials s Science ce Mark E. Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Sciences New York University, 100 Washington Square East, NY 10003 NYU-ECNU Center for Computational Chemistry at NYU Shanghai 200062, China 纽约大学 - 华东师范大学计算化学中心在上海纽约大学 NEW YORK UNIVERSITY MRSEC
Crystallization and the Pharmaceutical Industry: Materials Science? Pharmaceutical Materials Science: An Active New Frontier in Materials Research J. Elliott and B. Hancock Guest Editors MRS Bulletin 31 , Issue 11 (2006) Pharmaceutical Industry • $500 billion global market • $250 billion US market • Majority of pharmaceuticals are small molecule crystals “One of the continuing scandals in the physical sciences is that it remains impossible to predict the structure of even the simplest crystalline solids from their chemical composition.” -- John Maddox, Nature (1998)
Targets of the sixth blind structure prediction test
Accuracy of intermolecular interactions Standard force fields q q a b ( ) ( ) i j ij ij U R U R bonded 12 6 r r r ij ij ij ij Kohn-Sham density functional theory (plus dispersion): 1 1 ( ) ( ) n r n r min 2 ( ) [ ] [ , ] ( ) U R d d r r E n E n R U R 2 i i 2 ' xc ext NN r r { } i 2 ( ) ( ) r r n i i j ij i Empirical potentials fit to SAPT(DFT) calculations ( ) n q q C ( ) r (1) ( ) ( ) 1 ( , ) i j ( , ) ij ( ) l l n R R U A r e ij ij f r f r U 1 bonded ij ij ij ij n ij ij n r r 6,8,10 ij l n ij ij m n r ( , ) 1 r f r e n ! m 0 m Machine- learning (‘mathematical’) potentials
Structure prediction protocol for Target XXII Run MD at exptl. P,T Optimize monomer Generate Unit Cells geometry using DFT (UPACK) [PBE0 + D3, 6-311G**(d,p) 25,000 generated structures 4,042 unique after clustering 107 within 10 kJ/mol of lowest energy Lowest Energy Structures C2/c Pbca Pbca P2 1 2 1 2 1 P2 1 /n P2 1 /n
Summary of 50 predicted structures based on MD averaged energies
I: P2 1 /n II: Pbca, E = 3.46 kJ/mol III: P2 1 /c, E = 5.37 kJ/mol IV: P2 1 /n, E = 5.99 kJ/mol V: P2 1 /c, E = 6.31 kJ/mol
Prediction of the experimental structure Final ranking based on POLY1 SAPT(DFT) potential At 300 K : RMSD 20 from experimental structure = 0.18 Å. Rank = 4 on our list At 150 K : RMSD 20 from experimental structure = 0.15 Å. Rank = 4 on our list β Spc grp a b c P2 1 / n 11.95 6.70 12.60 108.6 Expt. P2 1 / n 12.02 6.72 12.48 108.7 Calc. Attempted predictions = 21, Times predicted = 12, Times ranked 5 or lower = 7
Pharmaceutical polymorphism Polymorphism refers to the ability of a compound to form multiple crystal structures. Ritonavir: HIV protease inhibitor • NIH: $3,500,000 investment • Abbott Labs: $200,000,000 investment • Originally dispensed in 1996 as ordinary capsules, no refrigeration required • Converted to lower energy (hitherto unknown) polymorph (form I to form II) on the shelf • Form II: Poor solubility, lower bioavailability • Required recall (1998) and reformulation as gel caps (2002). HIV-1 protease
Pharmaceutical Polymorphism C 13 H 22 N 4 O 3 S Ranitidine hydrochloride (Zantac) Acetylsalicylic acid Bond et al. Amer. Pharma. Rev . (2007)
Rough energy landscapes in proteins and crystals Dunitz and Scheraga PNAS 101 , 14309 (2004)
Sampling via collective variables in molecular simulation Peptides: φ ψ Crystals: Packing: Cell shape: 4 N l b 2 ( ) ( , ) Q f r Y l (2 1) b lm b b l N 1 m l b a b c x x x h Orientation: a b c y y y a b c z z z
Adiabatic free energy (temperature-accelerated molecular) dynamics (AFED/TAMD) L. Rosso, P. Minary, Z. Zhu, MET J. Chem. Phys . 116 , 4389 (2002) Margliano and Vanden-Eijnden, Chem. Phys. Lett. 426 , 168 (2006); J. B. Abrams and MET, J. Phys. Chem. B 112 , 14752 (2008) For prediction of crystal structures: Yu and MET Phys. Rev. Lett . 107 , 015701 (2011); Yu et al. J. Chem. Phys . 150 , 214109 (2014) Suppose n collective variables characterize a free energy landscape of interest ( ,..., ) ( ) 1,..., q q r r q r n 1 N Canonical probability distribution and free energy surface: n ( , ) ( ,..., ) d d ( ( ) ) H p r p r r P s s e q s 1 n 1 ( ,..., ) ln ( ,..., ) A s s k T P s s 1 1 n B n Write δ -functions as product of Gaussians: 1/2 2 ( ) lim exp ( ) q r s q r s 2 2 { } Dynamically sample Gaussian centers s 1 ,…, s n by introducing a kinetic energy for them And extending the phase space, which gives the following Hamiltonian: 2 p n n 2 ( , , , ) ( , ) s ( ) H p r p r r s p H q s s 2 2 m 1 1
Adiabatic free energy (temperature-accelerated molecular) dynamics (AFED/TAMD) Introduce high temperature for extended variables and high masses T T m m s i Adiabatically decoupled equations of motion: q H ( ) heat bath( ) r r m s q T i i r r i i ( ) heat bath( ) m s s q r T s ({ }) Under adiabatic conditions, we generate a distribution ( ,..., , , ) P s s T T adb 1 n s / ({ }) T T lim ( ,..., , , ) ( ,..., , ) s P s s T T P s s T adb 1 1 n s n { } ({ }) lim ln ( ,..., , , ) kT P s s T T adb 1 s n s { } T lim ln ( ,..., , ) ( ,..., , ) kT P s s T A s s T 1 1 s n n { } T s
Other developments: 1. Combining with metadynamics used as a bias: M. Chen. M. Cuendet, MET J. Chem. Phys. 137 , 024102 (2012) 2. Optimal free energy reconstruction protocols: M. Cuendet and MET J. Chem. Theor. Comput . 10 , 2975 (2014) Alanine Decamer (gas phase) Force field : CHARMM22 20 CVs : All ( φ , ψ ) pairs CV Temperature : T s = 900 K Physical Temp: T = 300 K CV mass : m ( ψ , φ ) = 600 m H Harmonic coupling : 300.5 kcal/mol/rad 2
Constant pressure version with cell matrix as CVs GROMOS FF, 100 K, 32,000 40,000 K T T s 216 (3 3 3), 2 GPa, Total run time = 500 ps, 5 ns N P
X-ray powder diffraction patterns Yonetani et al . (2001). II(98) II(01) 98 01
Free energies of benzene polymorphs Six-dimensional free energy surface analyzed using clustering based on a Gaussian mixture model. (see, also, J. Yang et al . Science 345 , 640 (2014)) P 2 1 / c Cmca C 2/ c P 4 1 2 1 2 P 2 1 / c MURI/ARO II(98) II(01)
Polymorphs of resorcinol • Resorcinol (C 6 H 6 O 2 ) is a simple benzene derivative. • At ambient conditions, forms two stable forms ( α and β ) depending on the relative orientations of the OH 1 . bonds, both having space group Pna 2 1 • High pressure phase ( δ phase) exists and arises from an α → δ at pressures above 3 GPa 2 . Structure unknown. • Another high pressure phase ( ε phase) can be reached from β phase above 5.6 GPa. This phase is also thought to be disordered based on Raman scattering 2 . • Another ambient polymorph ( γ phase) in the R b conformation recently found 3 . Its exact structure is unknown 4 . • Recent new experimental activity in study of resorcinol. 5 1 Robertson and Ubbelohde Proc. Royal Soc. London 167 122 (1938) 2 Rao et al. Phys. Rev. B 65 054108 (2002). 3 Kahr et al. Cryst. Growth Design 11 2070 (2011). 4 Zhu et al . Angew. Chem. Intl. Ed . (submitted) 5 J. Phys. Chem. B (2015); J. Mol. Struct . (2015).
polymorph
2 P 1
transition at 3 GPa
Preliminary free energy estimates for resorcinol Supercell length A form δ form a/A b/A c/A a/A b/A c/A Cell length 10.53 9.53 5.60 8.20 11.65 4.88 Cell angle 90 90 90 90 90 90 Space GP Pna2 1 Pna2 1
Energetic Co-crystals: DADP.TxTNB DADP TCTNB Polarizable system without pre-determined force field parameters = opportunity for ab initio enhanced sampling! Experimental Structures α β ( chloro- ) ( iodo- ) ( bromo- ) 1 3 Exp. Structures (Maztger Group): J. Am. Chem. Soc . 2015 , 137, 5074−5079 Angew. Chem. Int. Ed. 2013 , 52 , 6468-6471
Recommend


















![1 2 3 4 5 1. 6 1. Opioid growth factor ([Met5]enkephalin) prevents the incidence and retards](https://c.sambuz.com/37135/1-2-3-4-5-s.webp)




More recommend
Explore More Topics
Stay informed with curated content and fresh updates.