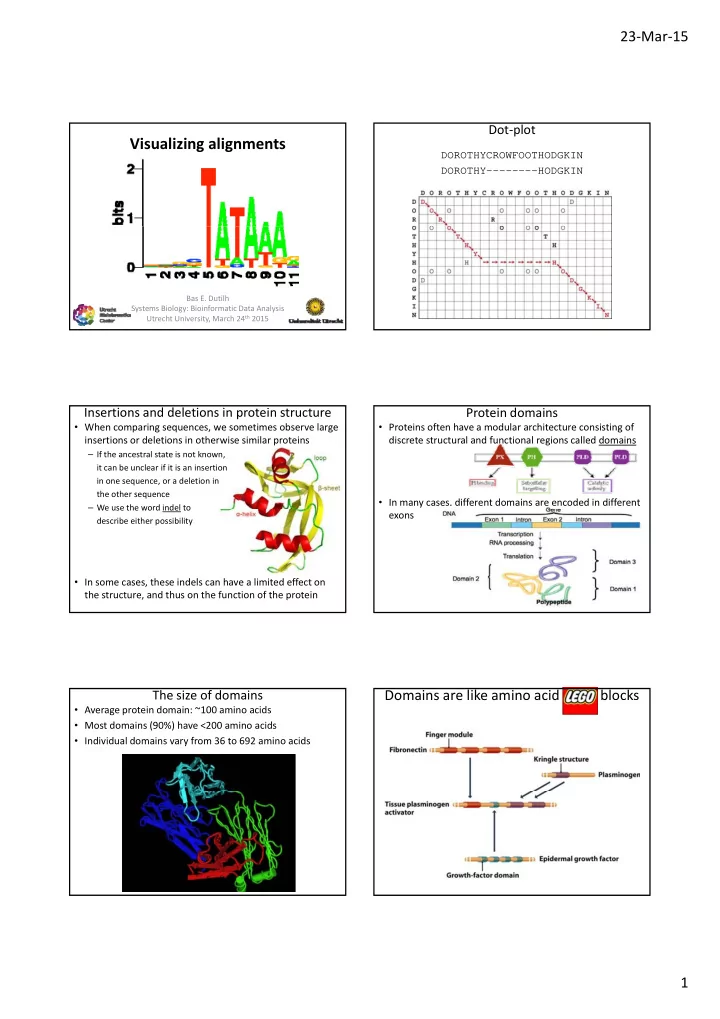
Visualizing alignments DOROTHYCROWFOOTHODGKIN - PDF document
23 Mar 15 Dot plot Visualizing alignments DOROTHYCROWFOOTHODGKIN DOROTHY--------HODGKIN Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 24 th 2015 Insertions and deletions in protein structure
23 ‐ Mar ‐ 15 Dot ‐ plot Visualizing alignments DOROTHYCROWFOOTHODGKIN DOROTHY--------HODGKIN Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 24 th 2015 Insertions and deletions in protein structure Protein domains • When comparing sequences, we sometimes observe large • Proteins often have a modular architecture consisting of insertions or deletions in otherwise similar proteins discrete structural and functional regions called domains – If the ancestral state is not known, it can be unclear if it is an insertion in one sequence, or a deletion in the other sequence • In many cases, different domains are encoded in different – We use the word indel to We use the word indel to exons describe either possibility • In some cases, these indels can have a limited effect on the structure, and thus on the function of the protein The size of domains Domains are like amino acid LEGO blocks • Average protein domain: ~100 amino acids • Most domains (90%) have <200 amino acids • Individual domains vary from 36 to 692 amino acids 1
23 ‐ Mar ‐ 15 Domain re ‐ arrangement can yield new proteins Discovering protein domains • By using domains, evolution can make new, complex • Protein domains can be discovered by using bioinformatics proteins! – Compare many protein sequences – Use local alignments – Src Homology 2 Colors allow you to visibly assess alignments Random unaligned sequences Well ‐ aligned homologs Color schemes Motifs in protein sequences • Color schemes are not always consistent • “Motif” rhymes with “beef” • DNA: each nucleotide has a different color • Motifs are functional units, but smaller A C G T • Proteins: different colors represent different physico ‐ than protein domains chemical properties of the amino acids • A motif is a short sequence pattern with a certain function – Some residues in the motif can be highly conserved in evolution • An alignment of occurrences of a motif shows which residues are more/less conserved in evolution – The active site of Hexokinase proteins 2
23 ‐ Mar ‐ 15 Motifs in DNA sequences Summarizing many aligned sequences • Inside gene coding regions: conserved genetic regions • Sometimes it is handy to summarize hundreds of aligned sequences • Outside coding regions: transcription factor binding sites (TFBS) • The consensus sequence is the sequence containing the most frequent residues at – TATA box: found in promoters of Archaea and Eukaryotes, binds transcription factors or histones each position – The TATA box • Unknown nucleotide: N • Unknown nucleotide: N • Unknown amino acid: X Sequence profiles Sequence profiles “summarize” biochemistry • Consensus of a protein motif of 4 amino acids: • A sequence profile represents all the possible sequences • A sequence profile shows much more detail: and the sequence conservation at once – Motifs Position 1 Position 2 Position 3 Position 4 A 0 0 0 – Protein families C 0 0 0 10 D 0 6 0 0 • This is almost like describing the real E 0 0 0 0 F 0 0 0 0 biochemical interactions! ...at G AA AT TT C ac... G 0 0 0 0 H 0 0 7 0 ...cc G AA GT TT C tg... g • We can predict that a conserved I 7 0 0 0 ...ag G AA AA TT C aa... K 0 0 0 0 ...gt G AA AT TT C cg... position is important for the function L 0 0 0 0 ...ca G AA AT TT C tc... M 0 0 0 0 of the protein because it is rarely ...tg G AA AT TT C gt... N 0 0 0 0 P 2 0 0 0 changed in evolution Q 0 0 0 0 R 0 0 0 0 A 2 1 0 6 6 5 1 0 0 0 2 1 S 0 4 0 0 • The profile shows: C 2 1 0 0 0 0 0 0 0 6 1 2 T 0 0 0 0 G 1 2 6 0 0 1 0 0 0 0 1 2 V 1 0 0 0 – Which positions are more conserved W 0 0 0 0 T 1 2 0 0 0 0 5 6 6 0 2 1 Most conserved position – Which positions are less conserved Y 0 0 3 0 DNA sequence logos Transcription factor binding sites (TFBS) At each position, possible nucleotides are shown by • Transcription factors are proteins that bind a specific DNA • stacked letters sequence – Letter heights relative to frequencies p i ( i = A , C , G , T ) The total stack height shows the conservation • – Information content at position k (in bits): The TATA box I ( k ) log ( 4 ) p log ( p ) 2 i 2 i i A , C , G , T Maximum information in a completely conserved position (e.g. always T ) • – p A = 0; p C = 0; p G = 0; p T = 1 – Assume that 0 log 2 (0) = 0 I = log 2 (4) + (0 + 0 + 0 + 1 log 2 (1)) = 2 Minimum information in a completely unconserved position (random) • – p A = 0.25; p C = 0.25; p G = 0.25; p T = 0.25 I = 2 + (0.25 log 2 (0.25) + 0.25 log 2 (0.25) + 0.25 log 2 (0.25) + 0.25 log 2 (0.25)) I = 2 + (– 0.5 – 0.5 – 0.5 – 0.5) = 0 3
23 ‐ Mar ‐ 15 Protein sequence logos An exam question Helix ‐ turn ‐ helix motifs I ( k ) log ( 4 ) p log ( p ) 2 i 2 i i A , C , G , T At each position, possible amino acids are shown by stacked letters • – Letter heights relative to amino acid frequencies p i ( p A , p C , p D , p E , p F , p G , p H , p I , p K , p L , p M , p N , p P , p Q , p R , p S , p T , p V , p W , and p Y ) I I ( ( k k ) ) log log ( ( 20 20 ) ) p p log log ( ( p p ) ) 2 2 i i 2 2 i i – The total stack height shows the conservation i 1 .. 20 – Information content at position k (in bits): a. Which positions are fully conserved? I ( k ) log ( 20 ) p log ( p ) 2 i 2 i i 1 .. 20 b. Which positions are fully random? Maximum information in a completely conserved position • c. Why is the y ‐ axis different between the two sequence logos? I = log 2 (20) + 0 = 4.3219 d. Give the maximum stack height for DNA sequence logos (in bits). Minimum information in a completely random position • e. Give the maximum stack height for protein sequence logos. I = 4.3219 + (20 · (– 0.216)) = 0 f. Give both the consensus sequences. Weblogo Useful programs • Weblogo is a webserver to create sequence logos from a • Bioinformatic programs to align sequences: multiple alignment: weblogo.berkeley.edu – Clustal – T ‐ Coffee – MAFFT • Programs to visualize alignments: – Clustal – Jalview – Seaview Jalview Weighing conservation of a position in an alignment Sequence identifiers Aligned sequences • Sequence alignments that use ...at G AA AT TT C ac... information about the sequence ...cc G AA GT TT C tg... conservation at each position into ...ag G AA AA TT C aa... ...gt G AA AT TT C cg... t G AA AT TT C account are called profile alignments ...ca G AA AT TT C tc... • In profile alignments, the important ...tg G AA AT TT C gt... (conserved) residues have a bigger impact on the alignment score – More conserved residues are weighed A 2 1 0 6 6 5 1 0 0 0 2 1 higher in the similarity score C 2 1 0 0 0 0 0 0 0 6 1 2 – Less conserved residues are weighed lower G 1 2 6 0 0 1 0 0 0 0 1 2 in the similarity score T 1 2 0 0 0 0 5 6 6 0 2 1 Conservation: identity at position → Pro fi le alignments are more sensi � ve than sequence alignments Quality: conservation of similar amino acids Consensus: frequency of top residue 4
Recommend






















More recommend
Explore More Topics
Stay informed with curated content and fresh updates.