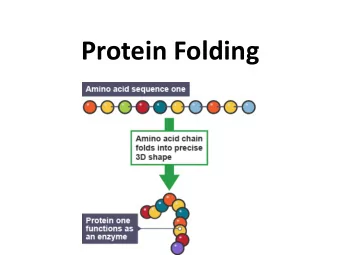
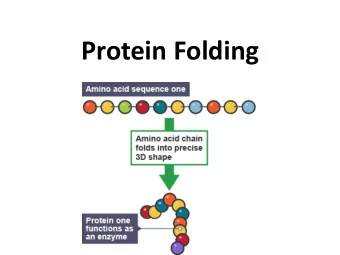
Summary Exploring modular protein architecture http://www.embl.de/gibson/SeqAnal/dev/wiki/index.php/Training/EmpaJan2010.html Wiki of the group of Toby Gibson at the EMBL in Heidelberg providing a number of presentations, tutorials and examples covering websites mentioned in the presentation and additional resources including (hoepfully) all the links Meta search engines and visualization tools http://srs.embl.de/srs/frontpage.do Query page for proteins, genes and related information allowing to search many pages at once http://biocompendium.embl.de/ BioCompendium is a publicly accessible, high-throughput experimental data analysis platform. It helps in prioritizing the targets from gene expression analysis studies, from RNAi studies or from similar experiments. http://reflect.ws/ Reflect highlights protein and small molecule names. To find out more about a highlighted term, just click on it. Can be run from the browser or by pasting an url of interest http://arena3d.org/ Arena3D introduces a new, staggered multi layer concept that allows the analysis of big networks in a three dimensional space representation. The different layers in the representation correspond to different data types respective concepts like sequences, structures, chemicals diseases, pathways etc. The data entries for one specific data type, like sequences, can be ordered or clustered on their respective layer by applying a data focused similarity measurement like sequence similarity. http://utopia.cs.man.ac.uk/ Utopia is a collection of interactive tools for analysing protein sequence and structure. Up front are user-friendly and responsive visualisation applications, behind the scenes a sophisticated model that allows these to work together and hides much of the tedious work of dealing with file formats and web services. Alignment tools and phylogenetic trees http://www.ebi.ac.uk/Tools/muscle/index.html
MUSCLE stands for MUltiple Sequence Comparison by Log-Expectation. MUSCLE is claimed to achieve both better average accuracy and better speed than ClustalW2 or T-Coffee, depending on the chosen options. http://www.ebi.ac.uk/mafft/ MAFFT (Multiple Alignment using Fast Fourier Transform) is a high speed multiple sequence alignment program. http://probcons.stanford.edu/ P ROB C ONS is an efficient protein multiple sequence alignment program, which has demonstrated a statistically significant improvement in accuracy compared to several leading alignment tools. http://molevol.cmima.csic.es/castresana/Gblocks_server.html Gblocks eliminates poorly aligned positions and divergent regions of a DNA or protein alignment so that it becomes more suitable for phylogenetic analysis. This server implements the most important features of the Gblocks program to make its use as simple as possible without loosing the functionality that it is necessary in most of the cases. http://bips.u-strasbg.fr/PipeAlign/Documentation/ PipeAlign is an on-line protein family analysis tool providing both interactive and automatic workbench for the validation, integration and presentation of the biological insights resulting from the analysis. It integrates a 5 step process ranging from the search for sequence homologues in protein sequence and 3D structure databases to the definition of the hierarchical relationship between and within subfamilies. Each step relies upon the results from the previous ones until a validated multiple alignment integrating subfamilies information is produced. The Pipe can also be started from any point and intermediate results are easily consulted. http://bips.u-strasbg.fr/fr/Products/Databases/BAliBASE2/ A benchmark alignment database, including enhancements for repeats, transmembrane sequences and circular permutations. Providing reference alignments http://www.treefam.org/ TreeFam (Tree families database) is a database of phylogenetic trees of animal genes. It aims at developing a curated resource that gives reliable information about ortholog and paralog assignments, and evolutionary history of various gene families http://eggnog.embl.de/version_2/ eggNOG (evolutionary genealogy of genes: Non-supervised Orthologous Groups) is a database of orthologous groups of genes. The orthologous groups are annotated with
functional description lines (derived by identifying a common denominator for the genes based on their various annotations), with functional categories (i.e derived from the original COG/KOG categories). http://pbil.univ-lyon1.fr/databases/hogenom/acceuil.php HOGENOM is a database of homologous genes from fully sequenced organisms (bacteria, archeaea and eukarya) , structured under ACNUC sequence database management system. It allows to select sets of homologous genes among species, and to visualize multiple alignments and phylogenetic trees. http://inparanoid.sbc.su.se/cgi-bin/index.cgi Addressed to the need to identify orthologs and paralogs http://phylomedb.org/?seqid=Q05609 PhylomeDB is a public database for complete collections of gene phylogenies (phylomes). It allows users to interactively explore the evolutionary history of genes through the visualization of phylogenetic trees and multiple sequence alignments. Moreover, phylomeDB provides genome-wide orthology and paralogy predictions which are based on the analysis of the phylogenetic trees. The automated pipeline used to reconstruct trees aims at providing a high-quality phylogenetic analysis of different genomes http://tardis.nibio.go.jp/homstrad/ OMSTRAD (HOMologous STRucture Alignment Database) is a curated database of structure- based alignments for homologous protein families. All known protein structure are clustered into homologous families (i.e., common ancestry), and the sequences of representative members of each family are aligned on the basis of their 3D structures using the programs MNYFIT, STAMP and COMPARER. These structure-based alignments are annotated with JOY and examined individually. Structural alignments http://fatcat.burnham.org/fatcat-cgi/cgi/fatcat.pl?-func=pairwise Comparing two protein structures by FATCAT. Users can provide two protein structures by uploading a pair of files in PDB format, or inputing a pair of PDB codes. FATCAT then calculates their structure alignment using either rigid or flexible comparison, depending on the users's choice. http://cl.sdsc.edu/ce/ce_align.html
Calculate structural alignment for two polypeptide chains either from the PDB or uploaded by the user http://topmatch.services.came.sbg.ac.at/TopMatchFlex.php?query=&target= PDB codes and chain identifiers of two protein structures to be compared. You can also directly access SCOP or CATH domains by entering the respective domain identifiers. Interactions http://stitch.embl.de/ STITCH is a resource to explore known and predicted interactions of chemicals and proteins. Chemicals are linked to other chemicals and proteins by evidence derived from experiments, databases and the literature. http://string.embl.de/ STRING is a database of known and predicted protein interactions. The interactions include direct (physical) and indirect (functional) associations; they are derived from four sources. STRING quantitatively integrates interaction data from these sources for a large number of organisms, and transfers information between these organisms where applicable. The database currently covers 2,590,259 proteins from 630 organisms. http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml CDD is a protein annotation resource that consists of a collection of well-annotated multiple sequence alignment models for ancient domains and full-length proteins. These are available as position-specific score matrices (PSSMs) for fast identification of conserved domains in protein sequences via RPS-BLAST. CDD content includes NCBI-curated domains, which use 3D-structure information to explicitly to define domain boundaries and provide insights into sequence/structure/function relationships, as well as domain models imported from a number of external source databases Disorder prediction http://globplot.embl.de/ Intrinsic Protein Disorder, Domain & Globularity Prediction http://www.disprot.org/ The Database of Protein Disorder (DisProt) is a curated database that provides information about proteins that lack fixed 3D structure in their putatively native states, either in their entirety or in part. http://iupred.enzim.hu/
Recommend
More recommend
Unleash a World of Digital Possibilities—Browse, Share, and Explore Content Without Boundaries