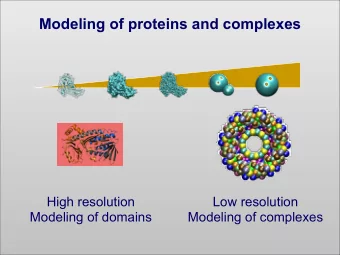
Molecular Modeling of Proteins: application to cancer immunotherapy O. Michielin (1,2,3) (1) Centre Pluridiscipinaire d'oncologie CHUV, Lausanne, Switzerland (2) Ludwig Institute for Cancer Research Epalinges, Switzerland (3) Swiss Institute of Bioinformatics Dorigny, Switzerland
Introduction & historical note Theoretical milestones: Classical equations of motion: F(t)=m a(t) Newton (1643-1727): Schrödinger (1887-1961): Quantum mechanical equations of motion: - ih ∂ t Ψ (t)=H(t) Ψ (t) Boltzmann(1844-1906): Foundations of statistical mechanics Molecular dynamics milestones: Metropolis (1953): First Monte Carlo (MC) simulation of a liquid (hard spheres) Wood (1957): First MC simulation with Lennard-Jones potential Liquids Alder (1957): First Molecular Dynamics (MD) simulation of a liquid (hard spheres) Rahman (1964): First MD simulation with Lennard-Jones potential Karplus (1977) & First MD simulation of proteins McCammon (1977) Proteins Karplus (1983): CHARMM general purpose FF & MD program Kollman(1984): AMBER general purpose FF & MD program Car-Parrinello(1985): First full QM simulations Kollmann(1986): First QM-MM simulations
Molecular Modeling Principles 1) Modeling of molecular interactions Electrostatics Van der Waals Covalent bonds Solvent Free energy landscape 2) Simulation of time evolution (Newton) 3) Computation of average values Connection microscopic/ O = < O > Ensemble = < O > Temps (Ergodicity) macroscopic Macroscopic value Average simulation value
Connection micro/macroscopic: intuitive view E 1 , P 1 ~ e - β E1 E 2 , P 2 ~ e - β E2 Expectation value O = 1 � E 3 , P 3 ~ e - β E3 O i e � � Ei Z i � e � � Ei Z = Where i E 4 , P 4 ~ e - β E4 is the partition function E 5 , P 5 ~ e - β E5
Central Role of the Partition Function � Z = e � � Ei i O = 1 � O i e � � Ei . . . Z i Expectation Value E = � � ln( Z ) � � ln( Z ) = U p = k B T G = -k B T ln (Z) � � � � � V � � N , T Internal Energy Pressure Gibbs free energy
Dynamical aspects of molecular recognition
Free energy: classical definition + The free energy is the energy left for once you paid the tax to entropy: � G = � H � T � S Enthalpic Entropic ● Hydrogen bonds ● Loss of degrees of freedom ● Polar interactions ● Gain of vibrational modes ● Van der Waals interactions ● Loss of solvent/protein structure ● ... ● ... Theoretical Predictions: ● Approximate: empirical formula for all contributions ● Exact: using statistical physics definition of G
Free energy: computational approaches � � � G = G A � G B = � k B T ln Z A � � � � Z B Free energy simulations techniques aim at computing ratios of partition functions using various techniques. � e � � Ei Z = i Sampling of important Computation of energy microstates of the system of each microstate (MD, MC, GA, …) (force fields, QM, CP, …)
The CHARMM Force Field ( b � b 0 ) 2 + � � V = ( � � � 0 ) 2 K b K � Bonds Angles � + ( � � � 0 ) 2 K � Impropers � [ ] + 1 � cos( n � � � � � ) K � Dihedrals q i q j � 1 + 4 �� r i > j i , j ij ) 12 � ( � ij / r [ ] � + 4 � ij ( � ij / r ij ) 6 i > j
Ergodic Hypothesis MD Trajectory NVT simulation E NVE simulation ψ ϕ 3 N Spatial coordinates “Alanine” Protein � ? O Ensemble = 1 d � d � = 1 � � O ( � , � ) e � � E ( � , � ) = O Time O ( t ) dt � Z 0
Free energy calculation: Main approaches Sampling, Exact Free Energy Perturbation (FEP) Thermodynamical Integration (TI) Non Equilibrium Statistical Mechanics (Jarzynski) CPU Time Sampling, Approx. Linear Interaction Energy (LIE) Molecular Mechanics/Poisson- Boltzmann/Surface area (MM-PBSA) Approx. Quantitative Structure Activity Relationship (QSAR) � G = F ( X ) G k 0 k i X i ( X is a descriptor)
Medical background: Cytotoxic activity of T lymphocytes T Lymphocyte Tumor Cell
Tumor cell recognition by CD8+ T cells: the TCR-p-MHC complex Lym phocyte CD8 + T Lym phocyte CD8 + T Lym phocyte X-ray X-ray structure of structure of Tum or cell bound TCR-p-MHC Tum or cell bound TCR-p-MHC
Goals of the molecular modeling approach Peptide vaccine TCR sequence optimization optimization Optimized peptide Adoptive vaccination Immunotherapy Clinical Trials
Principles of peptide based immunotherapy Peptide Injected Sub-Cutaneously (with Adjuvant) Peptidases Displacement
Regression of pulmonary melanoma metastases after vaccination with Melan-A peptide (patient LAU 446) July 9, 2001 September 24, 2001 < 0.1 % of Melan-A specific 0.3 % of Melan-A specific CD8+ T cells in PBL CD8+ T cells in PBL
Immunotherapy using adoptive transfert T lymphocytes extraction Transfection of Optmized TCR (viral vector) in vitro expansion Reinfusion (optionnal)
Lymphodepletion combined with adoptive transfert Dudley & al, JCO 2005
Goals of the molecular modeling approach Peptide vaccine TCR sequence optimization optimization Optimized peptide Adoptive vaccination Immunotherapy Clinical Trials
Free energy calculations: Absolute binding K A free energies: Δ G + → K A K D Relative binding e - Δ G/RT = K A free energies: ΔΔ G → K A’ / K A Free Energy Association Constant Binding free energy profiles: Δ G( ξ ) → K A , K on , K off Microscopic Structure Biological function
Free energy calculation: Main approaches Sampling, Exact Free Energy Perturbation (FEP) Thermodynamical Integration (TI) Non Equilibrium Statistical Mechanics (Jarzynski) CPU Time Sampling, Approx. Linear Interaction Energy (LIE) Molecular Mechanics/Poisson- Boltzmann/Surface area (MM-PBSA) Approx. Quantitative Structure Activity Relationship (QSAR) � G = F ( X ) G k 0 k i X i ( X is a descriptor)
Binding free energy decomposition: MM-PBSA, MM-GBSA Averaged over an MD simulation trajectory � E gaz of the complex (and isolated parts) Gaz Lig + Prot Lig:Prot � G bind = � E gaz + � G desolv � T � S � G solv � G solv � G solv lig prot comp E gaz = E elec + E vdw + � E int ra comp � � G solv lig + � G solv ( ) � G desolv = � G solv Sol Lig:Prot prot Lig + Prot Δ G bind � T � S = � T ( S comp � ( S prot + S lig )) S = S trans + S rot + S vib B. Tidor and M. Karplus, J. Mol. Biol ., 1994 , 238 , 405 � G solv = � G solv , elec + � G solv , np ( ) ) + � SASA comp � SASA lig + SASA prot ( ( ) � G desolv = � G solv , elec � � G solv , elec + � G solv , elec comp lig prot Depending on the way Δ G solv,elec is calculated: Molecular mechanics – Poisson-Boltzmann Surface Area (MM- PBSA) J. Srinivasan, P.A. Kollmann et al ., J. Am. Chem. Soc ., 1998 , 120 , 9401 Molecular mechanics – Generalized Born Surface Area (MM- GBSA) H. Gohlke, C. Kiel and D.A. Case, J. Mol. Biol ., 2003 , 330 , 891
MM-GBSA Method: application to TCR-p-MHC � G bind = � E gaz + � G desolv � T � S
Examples of TCR optimization: 2C TCR
Free energy calculation: Main approaches Sampling, Exact Free Energy Perturbation (FEP) Thermodynamical Integration (TI) Non Equilibrium Statistical Mechanics (Jarzynski) CPU Time Sampling, Approx. Linear Interaction Energy (LIE) Molecular Mechanics/Poisson- Boltzmann/Surface area (MM-PBSA) Approx. Quantitative Structure Activity Relationship (QSAR) � G = F ( X ) G k 0 k i X i ( X is a descriptor)
Computation of absolute TCR binding free energy Let G be the free energy and W the work, Pull W adia = Δ G K A (Infinitely slow) W = W adia + W diss (Finite rate) e � � W = e � � � G (Jarzynsky) Pull d W V C Independent starting pts (canonical ensemble) reaction reference trajectory time
Simulation setup - Gromos96 Force Field - Gromacs Engine - Particle Mesh Ewald (PME) - Periodic boundary conditions - Box: 80x80x150 A - 26000 Water molecules - 85000 Atoms - Hydrogen shaken - 2 fs timestep - 0.5 ns / 24h on 4 alpha CPU
TCR binding free energy profile: e � � W = e �� G
Application to the design of small molecule inhibitors EADock
Conformational sampling using genetic algorithms Final solution Angle 1 2 3 4 5 6 7 8 Value -139 -140 172 128 -23 137 175 -174 Optimized conformation (end of evolutionary cycle) Each selected Degree Of Freedom (DOF) Parents face an evolutionary process (The values of all DOFs is called a chromosome ) Select conformation with best fitness (lowest E) Angle 1 2 3 4 5 6 7 8 Value -92 -126 -138 -50 133 -125 -118 -144 Change DOF (Mutations, Angle 1 2 3 4 5 6 7 8 Population recombinations) Value 82 46 -40 -38 -46 -56 -134 -21 ... Angle 1 2 3 4 5 6 7 8 Replace worst Value -103 40 -139 6 106 -100 30 154 conformations The population is composed of a Offspring large number of chromosomes Seeding Evolutionary cycle
Eadock: Evolutionary Parameters Genome Cartesian coordinates Enthalpy Fitness Enthalpy & solvation free energy (GB, PB) Binding free energy … Rotations Operators Translations Followed by ElectrostaticOptimizer minimization VanDerWaalsOptimizer Barbatruc LigandInterpolator Automated Dihedral scan Operator scheduling Molecular Dynamics (SA) …
Recommend
More recommend
Unleash a World of Digital Possibilities—Browse, Share, and Explore Content Without Boundaries