
Metagenomics an introduction Katie Lennard Metagenomics vs. - PowerPoint PPT Presentation
Metagenomics an introduction Katie Lennard Metagenomics vs. amplicon sequencing (16S) Metagenomics = Shotgun sequencing of DNA from an environment Improved taxonomic resolution compared to 16S Depending on the 16S region, some
Metagenomics – an introduction Katie Lennard
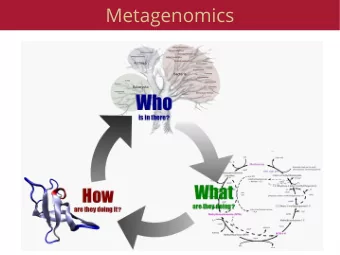
Metagenomics vs. amplicon sequencing (16S) • Metagenomics = Shotgun sequencing of DNA from an environment • Improved taxonomic resolution compared to 16S • Depending on the 16S region, some taxa are impossible to resolve • Limited to analysis of previously identified taxa • Offers compositional (who is there) AND functional (what are they doing) information • In the past few years metagenomic sequencing has been used to • Identify novel viruses • Characterize genomic diversity and function of uncultured bacteria • Reveal novel proteins • Identify taxa and metabolic pathways that differentiate disease states
Metagenomics analyses challenges • More expensive and computationally demanding • Require high performance compute clusters and parallelization • More complex pipeline that needs to be tailored to study • The challenge of correct binning – which read belongs to which organism? • For highly diverse communities (soil, gut) – coverage may be insufficient to characterize low abundance organisms • Host DNA (human, plant etc.) needs to be removed – mostly simple but may require PCR-based enrichment of microbial DNA if majority of DNA is host-derived (e.g. certain human samples; plants) • Contaminant removal tricky – which genes were generated by the contaminant?
Sequencing technologies overview
Common metagenomic techniques: marker gene analysis • Marker gene analysis (marker gene database) • Straightforward + computationally efficient • Apply to assembled OR unassembled reads • Two strategies • sequence similarity to marker gene database + custom classifiers that consider rate of evolution and read properties (MetaPhyler; MetaPhlAn) • based on phylogenetic information: identify metagenomic homologs of phylogenetically informative, single copy protein-coding genes (AMPHORA – Hidden Markov Models) → assemble a marker gene phylogeny (phylotyping)
Common metagenomic techniques: assembly • Crucial step for: • Genome reconstruction of individual organisms AND • To elucidate taxonomic and functional diversity of the community • Challenges: • Repeat regions • Co-assembly of reads from different (related) taxa → chimeric contigs • All assemblers will make numerous errors! • Manual inspection (time)
Common metagenomic techniques: binning • Reference-based (taxonomy dependent) • caveat: 3 million of 16S rRNA genes already sequenced, BUT only around 6000 complete genomes available • De novo (taxonomy independent) • Sequence composition based (e.g. %GC content) – need longer reads for accurate results (contigs); not reliable for complex microbial populations with low abundant communities. T • Abundance based – uses short (raw) reads or assembled contigs (method dependent) • Hybrid (sequence+abundance based) • Result used not only for taxonomic assignment but also downstream (genome assembly, evaluate functional profiles for each bin)
Common metagenomic techniques: gene prediction • Functional (what are they doing?) • Gene prediction (gene calling) – ID coding regions • Evidence-based (sequence similarity to database gene sequences) • ab initio (relies on intrinsic factors in DNA sequence to discriminate coding/non-coding) • Functional annotation
http://envgen.github.io/metagenomics.html
Self study resources • Sedlar et al. Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics Computational and Structural Biotechnology Journal 15 (2017) 48–55 • Ghurye et al. Metagenomic Assembly: Overview, Challenges and Applications https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5045144/ • Mande et al. Classification of metagenomic sequences: methods and challenges https://academic.oup.com/bib/article/13/6/669/193900/Classificatio n-of-metagenomic-sequences-methods
Recommend






















More recommend
Explore More Topics
Stay informed with curated content and fresh updates.