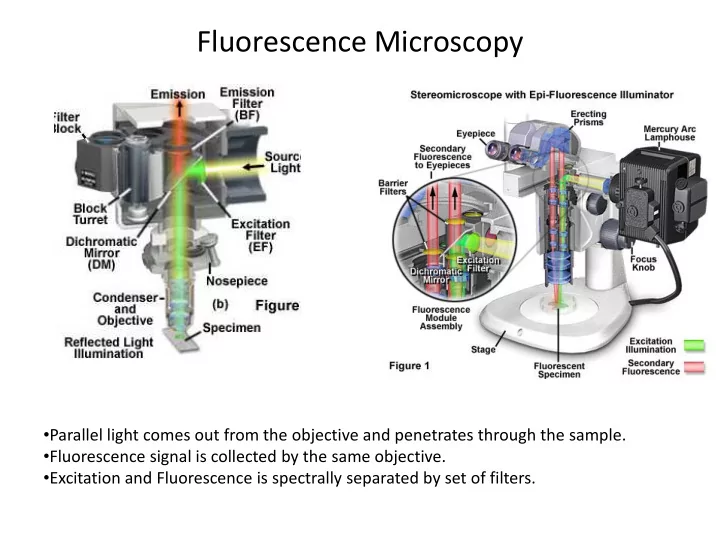
Fluorescence Microscopy Parallel light comes out from the objective - PowerPoint PPT Presentation
Fluorescence Microscopy Parallel light comes out from the objective and penetrates through the sample. Fluorescence signal is collected by the same objective. Excitation and Fluorescence is spectrally separated by set of filters.
Fluorescence Microscopy • Parallel light comes out from the objective and penetrates through the sample. • Fluorescence signal is collected by the same objective. • Excitation and Fluorescence is spectrally separated by set of filters.
Matching the Excitation with Fluorophore Absorption
Standard Filter Sets • If a lamp is used for excitation, narrow-band (10-20 nm) excitation filter is used to generate monochromatic light. • Laser emission is reflected from dichroic mirror and sent to the sample. • Fluorescence signal from the specimen is transmitted by the dichroic and filtered by emission filter.
Filter Design
Broad absorption and emission spectrum of fluorophores causes bleed-through between fluorescence channels http://www.invitrogen.com/site/us/en/home/support/Research-Tools/Fluorescence- SpectraViewer.html
Multi-Color Imaging EMITTER MIRROR Sequential excitation with laser lines with multi-band emission and dichroic filter set.
Wide-Field (Epi) Fluorescence Microscopy • A bright light source is used to excite fluorophores in the specimen. • For efficient high -contrast imaging, both the excitation illuminator and objective lens are positioned on the same side of the specimen. • In epi-illuminator, and the objective lens functions both as the condenser, delivering excitatory light to the specimen, and as the objective lens, collecting fluorescent light and forming an image of the fluorescent object in the image plane. • Fluorescence filter sets are positioned in the optical path between the epi-illuminator and Fluorescence signal the objective. • High -NA, oil immersion objectives made of low-fluorescence glass are used to maximize light collection and provide the greatest possible resolution and contrast
Epi Fluorescence Images human medulla rabbit muscle fibers sunflower pollen grain • Images are bright, but blurry and low contrast • Epi-illuminator excites the whole thickness of the specimen, unable to control the depth of the field • Bright fluorescent signal from out-of-focus objects give low-contrast • Autofluorescence (fluorescence signal from unlabeled objects) of the cell increases the background
Causes of High Fluorescent Background • Less then ideal filter sets. • Nonspecific binding of fluorophores to specimen. • Reflections and scattering in the optical pathway. • Dust and fingerprints in optics. • Fluorescence from other objects, including Raman scattering of water, autofluorescence of objective glass. • In multifluorescence applications, fluorescent bleedthrough between the channels.
Sources Autofluorescence in Cells Autofluorescence of Horsetail Fern (Plant Cell) Chlorophyll and polyphenols (in plant cells) Flavins (FDH), pyridine nucleotides (NADH) Pigments, serotonin Elastin, fibrilin Aromatic amino acid side chains (Trp, Phe)
Scanning Confocal Microscopy A laser point source is confocal with a scanning point in the specimen. Fluorescent wavelengths emitted from a point in the specimen are focused as a confocal point at the detector pinhole. Fluorescent light emitted at points above and below the plane of focus of the objective lens is not confocal with the pinhole and forms extended disks in the plane of the pinhole. Since only a small fraction of light from out-of- focus locations is delivered to the detector, out-of-focus information is largely excluded from the detector and final image.
Components of a Confocal Microscope • Epi illumination is used. • A laser beam is expanded to fill the back aperture of the objective and forms an intense diffraction-limited spot. • The pinhole aperture accepts fluorescent photons from the illuminated focused spot • Image is formed by raster scanning of the focused laser beam. • Magnification is generated by scanning step size. • Fluorescence signal is detected by single channel detector PMT. • Digital pixels are generated by analog to digital converter.
• Superior image contrast and clarity • Three dimensional view. • z stacks, yz and xz cross sections. • Five-dimensional views including information in x-, y-, and z-dimensions, in a timed sequence, and in multiple colors.
3D Scanning Confocal Microscopy • The primary advantage of laser scanning confocal microscopy is the ability to serially produce thin (0.5 to 1.5 micrometer) optical sections through fluorescent specimens • Objective can be moved in z direction with a piezoelectric motor to take z stack images. See tutorial in http://www.microscopyu.com/tutorials/java/virtual/confocal/index.html
Image Reconstruction The mouse intestine section The pollen grain 45 z stack reconstitution exterior surface
Photo Multiplier Tube highly sensitive photon detectors that do not require spatial discrimination, but instead respond very quickly with a high level of sensitivity to a continuous flux of varying light intensity
Analog to Digital Conversion
Sampling Frequency
Resolution in Confocal Microscopy In wide-field fluorescence optics, spatial resolution is determined by the wavelength of the emitted fluorescent light (left). In confocal mode, both the excitation and emission wavelengths are important, because the size of the scanning diffraction spot inducing fluorescence in the specimen depends directly on the excitation wavelength. Therefore, the volume both illuminated and observed is simply the product of two point spread functions (right).
Axial and Lateral Excitation The smallest distance that can be resolved using confocal optics is proportional to (1/ λ exc + 1/ λ em ) , and the parameters of wavelength and numerical aperture figure twice into the calculation for spatial resolution. Spatial resolution also depends on the size of the pinhole aperture at the detector, the zoom factor, and the scan rate.
• Decreasing the pinhole size reduces the thickness of the focal plane along the z-axis, thereby allowing higher resolution in optical sectioning. • Decreasing the pinhole size also improves contrast by excluding out-of-focal-plane light. • The lateral spatial resolution in the xy plane obtainable in a confocal fluorescence microscope can exceed that obtainable with wide-field optics by a factor of 1.4 . • Normally the detector pinhole is adjusted to accommodate the full diameter of the diffraction disk. • However, if the pinhole is stopped down to ¼ of the diffraction spot diameter , the effective spot diameter is slimmed down so that the disk diameter at one-half maximum amplitude is reduced by a factor of 1.4. minimum resolvable distance d x,y ~ 0.4 λ /NA d z ~ 1.4 λ n/NA 2
Image Quality and Performance Dynamic Range: Analog-to Digital Converter should be adjusted that minimum and maximum signal cover the whole dynamic range of the digitizer. Signal to Noise Ratio: [I/(I+B) 1/2 ] Background is generated by photon shot noise, detector readout, and dark-current Temporal Resolution : (Number of pixels X Dwell Time) per image Usually 1 µ sec per pixel and 512x512 pixels Optimum Pixel Size: ~ d /2 (smaller pixels are better when signal level is too high)
Image Optimization Total number of photons obtained from specimen is roughly constant! We need to use our photons economically for low signal levels. Intensity Spatial Resolution Pinhole Size + - Zoom + + Scan Rate - - Temporal Resolution Objective NA + + Laser Power + + Photobleaching
Bleed-Through Correction Post Image Analysis
Narrowing emission bands reduces the bleed through but also reduces the signal Sequential image acquisition can be used if excitation wavelength are distinct
http://www.olympusmicro.com/primer/java/confocalsimulator/index.html
Imaging Thick Specimens
Two Photon Confocal Microscopy • Illumination with intense 800 nm light can excite a fluorophore that is normally excited at 400 nm • Two photons must be absorbed simultaneously • Absorption efficiency is proportional to I 2 of the laser.
• Pulsed infrared lasers (Ti-Sapphire) compress photons in time domain and effectively increase the intensity for 2-photon absorption. • Excitation occurs only in focus, no pinhole is required to exclude out of focus fluorescence. • The use of near IR permits examination of thick specimens, up to 0.5 mm.
Imaging Thick Specimens • Absence of out-of-focus absorption allows more of the excitation light photons to reach the desired specimen level. • The red and infrared light employed in two-photon excitation undergoes less scattering than shorter wavelengths. • The effects of light scattering are less detrimental to two-photon microscopy than to confocal microscopy.
Imaging Thick Specimens
Recommend





















More recommend
Explore More Topics
Stay informed with curated content and fresh updates.