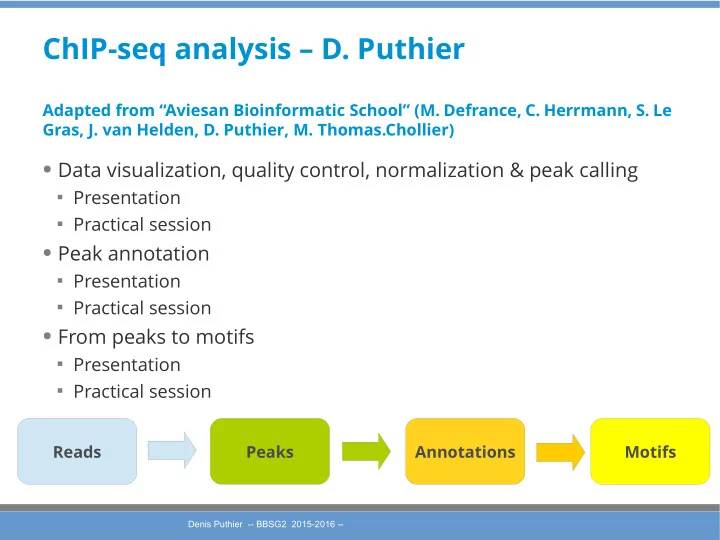
ChIP-seq analysis – D. Puthier Adapted from “Aviesan Bioinformatic School” (M. Defrance, C. Herrmann, S. Le Gras, J. van Helden, D. Puthier, M. Thomas.Chollier) ● Data visualization, quality control, normalization & peak calling Presentation Practical session ● Peak annotation Presentation Practical session ● From peaks to motifs Presentation Practical session Reads Peaks Annotations Motifs Denis Puthier -- BBSG2 2015-2016 -- Denis Puthier -- BBSG2 2015-2016 --
About transcriptional regulation and epigenetics Denis Puthier -- BBSG2 2015-2016 -- Denis Puthier -- BBSG2 2015-2016 --
A model of transcriptional regulation Denis Puthier -- BBSG2 2015-2016 --
Chromatin constraints ● E a c h d i p l o i d c e l l c o n t a i n s a b o u t 2 m e t e r s o f D N A H i g h l e v e l o f c o m p a c t i o n r e q u i r e d Accessibility required R e p l i c a t i o n Transcription DNA repair ● Specifjc machinery required Denis Puthier -- BBSG2 2015-2016 --
Chromatin has highly complex structure with several levels of organization Denis Puthier -- BBSG2 2015-2016 -- 2005. Genetics: A Conceptual Approach, 2nd ed.
Beads on a string F i g u r e 4 : C h r o m a t i n fj b e r s p u r i fj e d f r o m c h i c k e n e r y t h r o c y t e s . E a c h n u c l e o s o m e ( ~ 1 2 - 1 5 ● nm) is well resolved, along with the linker DNA between the nucleosomes. Given the resolution, other components, if present, such as a transcribing RNA polymerase or transcription factor complexes, should be resolvable Denis Puthier -- BBSG2 2015-2016 --
Histones and nucleosomes H i s t o n e s ● Small proteins (11-22 kDa) Highly conserved Basic (Arginine et Lysine) N-terminal tails subject to post translational modification Nucleosome ● Octamers of histone ( H 2 A , H 2 B , H 3 , H 4 ) x 2 146bp DNA Denis Puthier -- BBSG2 2015-2016 --
Nucleosome structure Denis Puthier -- BBSG2 2015-2016 --
Histone post translational modifjcation ● Lysine acetylation ● Lysine methylation ● Arginine methylation ● Serine phosphorylation ● Threonine phosphorylation ● ADP-ribosylation ● Ubiquitylation ● Sumoylation ● ... Denis Puthier -- BBSG2 2015-2016 --
Some alternative modifjcations Denis Puthier -- BBSG2 2015-2016 --
The Brno nomenclature The nomenclature set out here was devised following the fjrst meeting of the Epigenome Network of Excellence (NoE), at the Mendel Abbey in Brno, Czech Republic. For this reason, it can be referred to as the Brno nomenclature. Denis Puthier -- BBSG2 2015-2016 --
Epigenetic ● Epigenetics involves genetic control by factors other than an individual's DNA sequence Histone modifjcations DNA methylation ● Epigenetic modifjcations may be inherited mitotically or meiotically Denis Puthier -- BBSG2 2015-2016 --
Epigenetic and cancer Denis Puthier -- BBSG2 2015-2016 --
Chromatine immuno-precipitation (ChIP) ● Used for: TF localization Histone modifjcations Denis Puthier -- BBSG2 2015-2016 --
ChIP-Seq method Denis Puthier -- BBSG2 2015-2016 --
ChIP-Seq: technical considerations ● Quality of antibodies: one of the most important factors ('ChIP grade') High sensitivity Fivefold enrichment by ChIP-PCR at several positive-control regions High specifjcity The specifjcity of an antibody can be directly addressed by immunoblot analysis (knockdown by RNA-mediated interference or genetic knockout) Polyclonal antibodies may be prefered Ofger the fmexibility of the recognition of multiple epitopes ● Cell Number Typically 1 × 10 6 (e.g, RNA polymerase II/histone modifjcations) 10 × 10 6 (less-abundant proteins) Denis Puthier -- BBSG2 2015-2016 --
ChIP-Seq: technical considerations ● Open chromatin regions are easier to shear Higher background signals Two solutions Isotype control antibodies Immunoprecipitate much less DNA than specifjc antibodies Overamplifjcation of particular genomic regions during the library construction step (PCR) Duplicate PCR Input Non-ChIP genomic DNA Better control Denis Puthier -- BBSG2 2015-2016 --
Datasets used ● estrogen-receptor (ESR1) is a key factor in breast cancer developement ● goal of the study: understand the dependency of ESR1 binding on presence of co-factors, in particular GATA3, which is mutated in breast cancers ● approaches: GATA3 silencing (siRNA), ChIP-seq on ESR1 in wt vs. siGATA3 conditions, chromatin profjling Denis Puthier -- BBSG2 2015-2016 -- Denis Puthier -- BBSG2 2015-2016 --
Datasets used ● ESR1 ChIP-seq in WT & siGATA3 conditions ( 3 replicates = 6 datasets) ● H3K4me1 in WT & siGATA3 conditions (1 replicate = 2 datasets) ● Input dataset in MCF-7 (1 replicate = 1 dataset) ● p300 before estrogen stimulation ● GATA3/FOXA1 ChIP-seq before/after estrogen stimulation ● microarray expression data, etc ... Denis Puthier -- BBSG2 2015-2016 -- Denis Puthier -- BBSG2 2015-2016 --
Data processing & fjle formats Denis Puthier -- BBSG2 2015-2016 --
Fastq fjle format H e a d e r Sequence + (optjonal header) Quality (default Sanger-style) @QSEQ32.249996 HWUSI-EAS1691:3:1:17036:13000#0/1 PF=0 length=36 GGGGGTCATCATCATTTGATCTGGGAAAGGCTACTG + =.+5:<<<<>AA?0A>;A*A################ @QSEQ32.249997 HWUSI-EAS1691:3:1:17257:12994#0/1 PF=1 length=36 TGTACAACAACAACCTGAATGGCATACTGGTTGCTG + DDDD<BDBDB??BB*DD:D################# Denis Puthier -- BBSG2 2015-2016 --
Sanger quality score S a n g e r q u a l i t y s c o r e ( P h r e d q u a l i t y s c o r e ) : M e a s u r e t h e quality of each base call B a s e d o n p , t h e p r o b a l i t y o f e r r o r ( t h e p r o b a b i l i t y t h a t t h e corresponding base call is incorrect) Qsanger= -10*log10(p) p = 0.01 <=> Qsanger 20 Quality score are in ASCII 33 Note that SRA has adopted Sanger quality score although original fastq fjles may use difgerent quality score (see: htup://en.wikipedia.org/wiki/FASTQ_format) Denis Puthier -- BBSG2 2015-2016 --
ASCII 33 S t o r i n g P H R E D s c o r e s a s single characters gave a simple and space effjcient encoding: Character ”!” means a quality of 0 Range 0-40 Denis Puthier -- BBSG2 2015-2016 --
Quality control for high throughput sequence data FastQC GUI / command line htup://www.bioinformatjcs.bbsrc.ac.uk/projects/fastqc ShortRead Bioconductor package Denis Puthier -- BBSG2 2015-2016 --
Trimming Depending on the aligner this step can be mandatory Tools FASTX-Toolkit Sickle Window-based trimming (unpublished) ShortRead Bioconductor package ... Denis Puthier -- BBSG2 2015-2016 --
Quality control with FastQC Quality Position in read Denis Puthier -- BBSG2 2015-2016 --
Quality control with FastQC Position in read Denis Puthier -- BBSG2 2015-2016 --
Quality control with FastQC Nb Reads Mean Phred Score Denis Puthier -- BBSG2 2015-2016 --
Mapping reads to genome: general sofuwares a Work well for Sanger and 454 reads, allowing gaps and clipping. b Paired end mapping. c Make use of base quality in alignment.dBWA trims the primer base and the fjrst color for a color read. e Long-read alignment implemented in the BWA-SW module. fMAQ only does gapped alignment for Illumina paired-end reads. Denis Puthier -- BBSG2 2015-2016 -- g Free executable for non-profjt projects only.
Bowtje principle U s e h i g h l y e ffj c i e n t c o m p r e s s i n g a n d m a p p i n g a l g o r i t h m s b a s e d o n B u r r o w s Wheeler Transform (BWT) The Burrows-Wheeler Transform of a text T, BWT(T), can be constructed as follows. The character $ is appended to T, where $ is a character not in T that is lexicographically less than all characters in T. The Burrows-Wheeler Matrix of T, BWM(T), is obtained by computjng the matrix whose rows comprise all cyclic rotatjons of T sorted lexicographically. acaacg$ $acaacg 1 7 BWT (T) T caacg$a aacg$ac 2 3 aacg$ac acaacg$ 3 1 gc$aaac acaacg$ acg$aca acg$aca 4 4 cg$acaa caacg$a 5 2 g$acaac cg$acaa 6 5 $acaacg g$acaac 7 6 Denis Puthier -- BBSG2 2015-2016 --
Bowtje principle Burrows-Wheeler Matrices have a property called the Last First (LF) Mapping. The ith occurrence of character c in the last column corresponds to the same text character as the ith occurrence of c in the fjrst column. Example: searching ”AAC” in ACAACG 7 3 1 4 2 5 6 Denis Puthier -- BBSG2 2015-2016 --
Storing alignment: SAM Format Store informatjon related to alignement Read ID CIGAR String Bitwise FLAG read paired read mapped in proper pair read unmapped, ... Alignment positjon Mapping quality ... Denis Puthier -- BBSG2 2015-2016 --
Bitwise fmag read paired read mapped in proper pair read unmapped mate unmapped read reverse strand mate reverse strand fjrst in pair second in pair not primary alignment read fails platgorm/vendor quality checks read is PCR or optjcal duplicate Denis Puthier -- BBSG2 2015-2016 --
Recommend
More recommend
Unleash a World of Digital Possibilities—Browse, Share, and Explore Content Without Boundaries