Gaussian Accelerated Molecular Dynamics (GaMD) Yinglong Miao Center - PowerPoint PPT Presentation
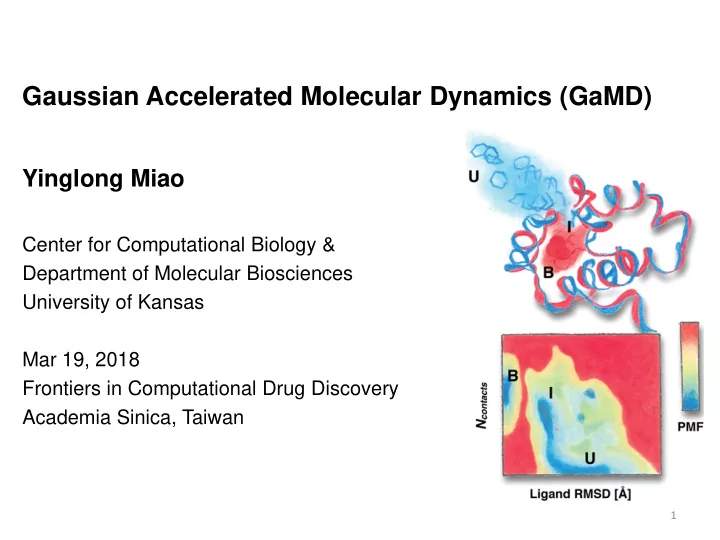
Gaussian Accelerated Molecular Dynamics (GaMD) Yinglong Miao Center for Computational Biology & Department of Molecular Biosciences University of Kansas Mar 19, 2018 Frontiers in Computational Drug Discovery Academia Sinica, Taiwan 1
Gaussian Accelerated Molecular Dynamics (GaMD) Yinglong Miao Center for Computational Biology & Department of Molecular Biosciences University of Kansas Mar 19, 2018 Frontiers in Computational Drug Discovery Academia Sinica, Taiwan 1
Outline Method: Gaussian Accelerated Molecular Dynamics (GaMD) Applications Protein folding and conformational sampling 1. Protein-ligand binding and unbinding 2. Protein-protein binding 3. Practical Usage of GaMD 2
Computer Simulation is Limited Large gap remains between simulation and biological timescales: This often leads to insufficient sampling and inaccurate free energy calculations of biomolecules: Protein functional free energy landscape 3
Enhanced Sampling is often Constrained Adaptive Biasing Force Umbrella Sampling o o Metadynamics Conformational Flooding o o Require predefined reaction coordinates, e.g., atom distances, torsion angles, etc. Abrams and Bussi, Entropy , 2014. 4
Accelerated Molecular Dynamics (aMD) Unconstrained enhanced sampling by adding a boost potential ΔV to reduce system energy barriers: V( r ) : Original potential energy ΔV(r) : Boost potential E : Reference energy α : Acceleration factor Hamelberg, Mongan and McCammon, J. Chem. Phys., 2004. 5
But large energetic noise in aMD ΔV ~ tens to hundreds of kcal/mol A long-standing problem in calculating free energy profiles from aMD simulations, notably for proteins! Shen and Hamelberg, J. Chem. Phys. , 2008. 6
How to achieve fast & accurate accelerated computer simulations, especially for large biomolecules like proteins? o Unconstrained enhanced sampling o Free energy calculation 7
Gaussian Accelerated Molecular Dynamics (GaMD) GaMD enhances sampling by reducing energy barriers with a harmonic boost potential, which follows Gaussian distribution: V( r ) : Original potential energy ΔV( r ) : Boost potential E : Reference energy k : Harmonic force constant. Miao , Feher and McCammon, J. Chem. Theory Comput. , 2015. 8
Optimal boost potential is added in GaMD 1. Keep overall shape of original potential energy surface 2. Reduce energy barriers for enhanced sampling 3. Set narrow distribution for the boost potential Miao , Feher and McCammon, J. Chem. Theory Comput. , 2015. 9
How to reweight GaMD simulations? Cumulant expansion to 2 nd order (“Gaussian Approximation”)*: • ΔV ~ 0 – 50 kcal/mol • Anharmonicity of ΔV Distribution is defined as : ΔV is dimensionless as divided by k B T 10 * Miao , Sinko, Pierce, Bucher, Walker and McCammon, JCTC , 2014.
GaMD Enables Accurate Free Energy Calculation Cumulant expansion to the 2 nd order is exact: ΔV ~ 0 – 50 kcal/mol F(A) : Original free energy; F*(A) : GaMD free energy; β = 1/k B T ; F c : Constant; C 1 , C 2 : Boost potential cumulants. Miao , Feher and McCammon, J. Chem. Theory Comput. , 2015. 11
Outline Method: Gaussian Accelerated Molecular Dynamics (GaMD) Applications Protein folding and conformational sampling 1. Protein-ligand binding and unbinding 2. Protein-protein binding 3. Practical Usage of GaMD 12
Protein Folding Chignolin: PDB GaMD
Protein Folding: Chignolin ΔV follows Gaussian distribution: ΔV avg (kcal/mol) 9.8 σ Δ V 2.4
Protein Folding: Free Energy Profile Folded (F), Intermediate (I) and Unfolded (U) states
Protein-Ligand Binding T4-lysozyme (protein) + Benzene (ligand): C terminal domain N terminal PDB: 181L domain 16
Protein-Ligand Binding Benzene binding to T4-lysozyme: ~50 μs ( k on = 8x10 5 − 10 6 M -1 S -1 )* Binding time: ~100 ns (GaMD); ~ 500x speedup * Feher, Baldwin and Dahlquist . Nat. Struct. Biol . 1996.
Protein-Ligand Binding ΔV follows Gaussian distribution: Δv avg (kcal/mol) 36.3 σ Δ V 4.7
Protein-Ligand Binding: Free Energy Profile Ligand RMSD : Relative to crystal structure; N contact : # of protein atoms in contact with ligand. GaMD quantitatively characterizes the Unbound (U), Intermediate (I) and Bound (B) states. 19
Protein-Ligand Binding HIV Protease: Semi-open Closed
GaMD Captured Ligand Binding to HIV Protease Miao, Huang, Walker, McCammon and Chang, Biochemistry , 2018 .
Comparison with Anton MD Simulation Miao, Huang, Walker, McCammon and Chang, Biochemistry , 2018 .
Ligand Binding: Free Energy Landscape Miao, Huang, Walker, McCammon and Chang, Biochemistry , 2018 .
Protein-Ligand Binding & Unbinding M 2 Muscarinic GPCR + Arecoline (~5 μM binding affinity): Drug pathway & binding sites Valuable information for drug design 24 Miao and McCammon, Proc. Natl. Acad. Sci. U. S. A., 2016.
Protein-Protein Binding M 2 Muscarinic GPCR + G-protein mimetic nanobody: 25 Miao and McCammon, Proc. Natl. Acad. Sci. U. S. A., 2018.
GaMD Advantages ~10 3 times faster than standard computer simulations Efficient: Unconstrained Enhanced Sampling Critical for simulating protein folding, ligand binding & unbinding, etc. Accurate: Free Energy Calculation Quantitative description of biomolecular dynamics. Available in widely-used AMBER, NAMD: http://miao.compbio.ku.edu/GaMD/ 26
Outline Method: Gaussian Accelerated Molecular Dynamics (GaMD) Applications Protein folding and conformational sampling 1. Protein-ligand binding and unbinding 2. Protein-protein binding 3. Practical Usage of GaMD 27
GaMD Implementation in AMBER Originally in Amber12 A patch was available for Amber 14 (“ amber14_GaMD_patch_confidential.txt ”): CPU: pmemd and pmemd.MPI o GPU: pmemd.cuda and pmemd.cuda.MPI o Officially released in AMBER16
GaMD Parameters Minimal set of input parameters: igamd Flag to apply boost potential = 0 (default) no boost is applied = 1 boost on the total potential energy only = 2 boost on the dihedral energy only = 3 dual boost on both dihedral and total potential energy irest_gamd Flag to restart GaMD simulation = 0 (default) new simulation = 1 restart simulation iE Flag to set the threshold energy E = 1 (default) set threshold energy to lower bound E = V max = 2 set threshold energy to upper bound E = V min +( V max - V min )/ k 0 ntcmd Number of cMD steps to calculate V max , V min , V avg , σ V (default 1,000,000) nteb Number of biasing equilibration steps (default 1,000,000) Upper limit of the standard deviation of total potential boost sigma0P (default 6.0 kcal/mol). sigma0D Upper limit of the standard deviation of dihedral potential boost (default 6.0 kcal/mol).
Example: Alanine Dipeptide How to run GaMD simulations with different dihedral-, total- and dual-boost potentials? How to analyze simulation trajectories? How to reweight GaMD simulations for free energy calculations?
GaMD: Alanine Dipeptide http://miao.compbio.ku.edu/GaMD/lib/GaMD_ Amber-Tutorial.pdf http://miao.compbio.ku.edu/GaMD/lib/amber- gamd-tutorial.tgz cp amber14-gamd-tutorial.tgz /data/ userid / cd /data/ userid /; tar -xvzf amber14-gamd- tutorial.tgz export GaMDHOME=/data/ userid /amber14- gamd-tutorial
Total-boost GaMD cd $GaMDHOME/test-dia/gamd-tot md.in &cntrl imin = 0, irest = 0, ntx = 1, nstlim = 1000, dt = 0.002, ntc = 2, ntf = 2, tol = 0.000001, iwrap = 1, ntb = 1, cut = 8.0, ntt = 3, temp0 = 300.0, tempi = 300.0, ntpr = 50, ntwx = 50, ntwr = 500, ntxo = 1, ioutfm = 1, ig = -1, ntwprt = 22, igamd = 1, iE = 1, irest_gamd = 0, ntcmd = 200, nteb = 200, ntave = 100, sigma0P = 6.0, &end qsub run-gamd.pbs pmemd.cuda -O -i md.in -o md-1.out -p dip.top -c dip.crd - r md-1.rst -x md-1.nc
Dihedral-boost GaMD cd $GaMDHOME/test-dia/gamd-dih md.in &cntrl imin = 0, irest = 0, ntx = 1, nstlim = 1000, dt = 0.002, ntc = 2, ntf = 2, tol = 0.000001, iwrap = 1, ntb = 1, cut = 8.0, ntt = 3, temp0 = 300.0, tempi = 300.0, ntpr = 50, ntwx = 50, ntwr = 500, ntxo = 1, ioutfm = 1, ig = -1, ntwprt = 22, igamd = 2, iE = 1, irest_gamd = 0, ntcmd = 200, nteb = 200, ntave = 100, sigma0D = 6.0, &end qsub run-gamd.pbs pmemd.cuda -O -i md.in -o md-1.out -p dip.top -c dip.crd - r md-1.rst -x md-1.nc
Dual-boost GaMD cd $GaMDHOME/test-dia/gamd-dual md.in &cntrl imin = 0, irest = 0, ntx = 1, nstlim = 1000, dt = 0.002, ntc = 2, ntf = 2, tol = 0.000001, iwrap = 1, ntb = 1, cut = 8.0, ntt = 3, temp0 = 300.0, tempi = 300.0, ntpr = 50, ntwx = 50, ntwr = 500, ntxo = 1, ioutfm = 1, ig = -1, ntwprt = 22, igamd = 3, iE = 1, irest_gamd = 0, ntcmd = 200, nteb = 200, ntave = 100, sigma0P = 6.0, sigma0D = 6.0, &end qsub run-gamd.pbs pmemd.cuda -O -i md.in -o md-1.out -p dip.top -c dip.crd -r md-1.rst -x md-1.nc
Dual-boost GaMD: Restart simulation cd $GaMDHOME/test-dia/gamd-dual md-restart.in &cntrl imin = 0, irest = 1, ntx = 5, nstlim = 1000, dt = 0.002, ntc = 2, ntf = 2, tol = 0.000001, iwrap = 1, ntb = 1, cut = 8.0, ntt = 3, temp0 = 300.0, tempi = 300.0, ntpr = 50, ntwx = 50, ntwr = 500, ntxo = 1, ioutfm = 1, ig = -1, ntwprt = 22, igamd = 3, iE = 1, irest_gamd = 1 , ntcmd = 0, nteb = 0, ntave = 100, sigma0P = 6.0, sigma0D = 6.0, &end qsub run-gamd-restart.pbs pmemd.cuda -O -i md-restart.in -o md-2.out -p dip.top -c md-1.rst -r md-2.rst -x md-2.nc
Recommend





















More recommend
Explore More Topics
Stay informed with curated content and fresh updates.