
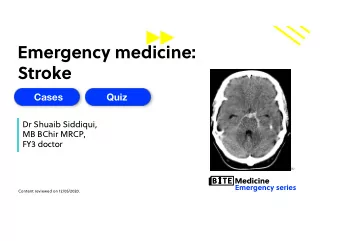
First-principle simulations of Zn-induced aggregation of -amyloid - PowerPoint PPT Presentation
First-principle simulations of Zn-induced aggregation of -amyloid peptides P. Giannozzi Universit` a di Udine and CNR-IOM Democritos, Trieste, Italy HPC User day: Aspettando Fermi CINECA, 7 maggio 2012 Work done in collaboration with:
First-principle simulations of Zn-induced aggregation of β -amyloid peptides P. Giannozzi Universit` a di Udine and CNR-IOM Democritos, Trieste, Italy HPC User day: Aspettando Fermi – CINECA, 7 maggio 2012 Work done in collaboration with: Karl Jansen, Francesco Stellato (DESY), Velia Minicozzi, Silvia Morante, Giancarlo Rossi (Roma II), Giovanni La Penna (ICCOM) – Typeset by Foil T EX –
Aggregation of peptides induced by metal ions Very nasty degenerative illnesses are caused by aggregation of naturally present proteins or peptides into toxic amyloid fibrils and plaques In Alzheimer disease, the main components of plaques are β -amyloids peptides (A β ): chains of 39 to 43 aminoacids, obtained by cleavage of a precursor protein (in the figure: A β 40 peptide in water) There is experimental evidence that transition metal ions Cu, Zn, Fe play a role in the processes of A β aggregation and plaque formation The details of the metal-A β binding are thus subject of intense study
β -amyloids binding with Cu and Zn: state of the art • The structure of A β binding with Cu is relatively well characterized, with Cu having a stable intra-peptide coordination • A β binding with Zn is not as clear. Competing structural models – from XAS: inter-peptide Zn 2+ bridge between three or more histidines (His) belonging to different peptides. Rather peculiar and infrequent: hallmark of peptide aggregation? – from NMR: intra-peptide binding to three His and either the N-terminus or a residue (Glu 11 ) • Competition for peptide binding between Cu and Zn ions likely Goal of this work: to find, using numerical simulations, realistic configurations for A β chains coordinated by Zn 2+ , fitting XAS results
Simulation procedure • Initial configurations generated with graphical tools (VMD), optimized with Amber force fields and Monte Carlo Random Walk • Selected configurations truncated (aminoacids 1-10 removed), optimized, set into an orthorhombic cell filled with water molecules, thermalized with classical MD, optimized with Tight-Binding MD • Finally, first-principle (i.e. from electronic structure) Car-Parrinello Molecular Dynamics runs are performed to check the stability and refine the structure of the various binding configurations The last step is by far the most time-consuming, requiring parallel execution on big computer facilities, including the BG/P (courtesy of DEISA DECI and of John von Neumann Institute for Computing)
Choosing the starting configurations Four good starting models (generated for A β 16 ) compatible with XAS data (many more turned out to be bad and were discarded): • S1: Zn bound to four histidines • S2: Zn bound to three histidines • S3: two Zn ions, bound to four histidines • S4: two Zn ions, bound to three peptides
First-principle (i.e. from electronic structure) simulations: Density-Functional Theory Transforms the many-electron problem into an equivalent problem of (fictitious) non-interacting electrons, the Kohn-Sham Hamiltonian : � � h 2 − ¯ 2 m ∇ 2 Hφ v ≡ r + V R ( � r ) φ v ( � r ) = ǫ v φ v ( � r ) � The effective potential is a functional of the charge density: Z I e 2 � � r ) | 2 V R ( � r ) = − + v [ n ( � r )] , n ( � r ) = | φ v ( � r − � | � R I | v I (Hohenberg-Kohn 1964, Kohn-Sham 1965). Exact form is unknown, but simple approximate forms yielding useful results are known. DFT is in principle valid for ground-state properties only.
Density-Functional Theory II The total energy is also a functional of the charge density: h 2 − ¯ � � � φ ∗ r ) ∇ 2 φ v ( � E ⇒ E [ φ, R ] = v ( � r ) d� r + V R ( � r ) n ( � r ) d� r + 2 m v � n ( � r ) n ( � e 2 e 2 r ′ ) Z I Z J r ′ + E xc [ n ( � rd� � + r )] + d� r − � | � R I − � 2 2 r ′ | | � R J | I � = J Kohn-Sham equations from the minimization of the energy functional: � φ ∗ E ( R ) = min φ E [ φ, R ] , i ( � r ) φ j ( � r ) d� r = δ ij Hellmann-Feynman theorem holds. Forces on nuclei: � � F I = −∇ � R I E ( R ) = − n ( � r ) ∇ � R I V R ( � r ) d� r
Plane-Wave Pseudopotential method • The introduction of pseudopotentials allows one to ignore chemically inert core states and to use a plane waves basis set • Plane waves are orthogonal and easy to check for completeness ; they allow to efficiently calculate the needed Hφ products and to solve the Poisson equation using Fast Fourier Transforms (FFTs) • Supercells allow to study systems in which perfect periodicity is broken (surfaces, defecs) or absent (amorphous, liquids) • Iterative techniques like Car-Parrinello Molecular Dynamics allow to treat rather big systems with affordable computational effort
Car-Parrinello Molecular Dynamics Introduce fictitious dynamics on the electronic orbitals φ v : r + 1 � | ˙ � � r ) | 2 d� M I ∇ 2 L = µ φ v ( � R I − E [ φ, R ] � 2 v I ( µ = fictitious electronic mass), subject to orthonormality constraints on the orbitals, implemented via Lagrange multipliers Λ ij . The above Lagrangian generates the following equations of motion: φ i = − δE M I ¨ µ ¨ � � + Λ ij φ j R I = −∇ � R I E [ φ, R ] δφ i ij (nuclear motion is classical). These equations can be integrated (i.e. solved) for both electrons and nuclei using classical Molecular Dynamics algorithms. The combined electronic and nuclear dynamics keeps electrons close to the ground state.
Technical details • Perdew-Burke-Erzerhof (PBE) exchange-correlation functionals • Ultrasoft (Vanderbilt) pseudopotentials with 25 Ry (orbitals) or 250 Ry (charge density) kinetic energy cutoff for plane waves • Simulation cell size: – S1: 1351 atoms, 21.291 × 35.193 × 23.241˚ A 3 – S2: 1204 atoms, 21.976 × 27.947 × 22.881˚ A 3 – S3: 1349 atoms, cell as S1 – S4: 2347 atoms, 32.083 × 31.01 × 23.14˚ A 3 : Tight-Binding MD only, too big for Car-Parrinello MD • At least 3.6 ps simulation time
Software: Quantum ESPRESSO Car-Parrinello simulations have been performed using the CP package of quantum ESPRESSO : Quantum opEn-Source Package for Research in Electronic Structure, Simulation, and Optimization . See: http://www.quantum-espresso.org for more info. quantum ESPRESSO is a distribution of software for atomistic calculations based on electronic structure, using density-functional theory, a plane-wave basis set, pseudopotentials. Freely available under the terms of the GNU General Public License. Main goals of quantum ESPRESSO are innovation in methods and algorithms; efficiency on modern computer architectures. quantum ESPRESSO implements multiple parallelization levels
Parallelization levels for CP group distributed quantities communications performances plane- PW, G -vector coefficients, high good CPU scaling, wave R -space FFT arrays good load balancing, distributes most RAM task FFT on electron states high improves load balancing linear- subspace hamiltonians very high improves scaling, algebra and constraints matrices distributes more RAM OpenMP FFT, libraries intra-node extends scaling on multicore machines A further parallelization level on electron states has since been added. MPI+OpenMP parallelization crucial for good BG/P performances
Mixed MPI-OpenMP scalability Two models of graphene on Ir surface on a BlueGene/P using 4 processes per computing node. Execution times in s , initialization + 1 self-consistency step. N cores T cpu (wall) T cpu (wall) 443 atoms 686 atoms 16384 740(772) 2861 (2915) 32768 441(515) 1962 (2014) 65536 327(483) 751 (1012) Fragment of an A β -peptide in water (the large difference between CPU and containing 838 atoms and 2312 wall time is likely due to I/O) A 3 cell, electrons in a 22.1 × 22.9 × 19.9˚ Γ -point. CP code on BlueGene/P, 4 processes per computing node.
Results Structures for S1, S2, S3 models before and after CP-MD. Note the fourth His leaving the Zn site in S1, while in S3 model Zn keeps a stable fourfold coordination
Results: a nice cover picture... Metallomics 4 , 156-165 (2012) (S4 model)
More serious results: simulated XAS spectra
Similar structures in other biological systems? Comparison of the Zn a site in S4 model (green: fit to XAS) with the Zn site in the reduced bovine superoxide-dismutase (SOD) enzyme Only residues involved in binding with Zn are shown
Discussion and conclusions • XAS yields information on short-range structure (up to 5 ÷ 6˚ A) only • First-principle techniques can take into account both the peculiar chemical binding of metals with peptides and the electrostatic interactions between peptides • Structures (S3 and S4) in which Zn is bound in a stable ways to four His have been identified... • ...but their structure is not trivial, requiring a second Zn and/or a third peptide chain; in the simpler S1 structure Zn loses a His and a satisfactory XAS fit is not obtained • S4 model reminiscent of the Zn site of bovine SOD
Recommend






















More recommend
Explore More Topics
Stay informed with curated content and fresh updates.