Bile acid abnormalities in peroxisomal disorders Sacha Ferdinandusse Lab. Genetic Metabolic Diseases Academic Medical Center Amsterdam
Peroxisomes play an important role in bile acid biosynthesis Bile acid biosynthesis involves: • Modification of the ring structure of cholesterol • Oxidation of the side chain • Shortening of the side chain------Peroxisome • Conjugation------Peroxisome
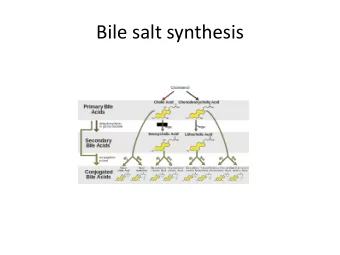
Peroxisomal C27-bile acid intermediates Cholesterol Ring modifications Side chain oxidation 3 α ,7 α -dihydroxycholestanoic acid 3 α ,7 α ,12 α -trihydroxycholestanoic acid (DHCA) (THCA) Side chain shortening via peroxisomal β -oxidation Chenodeoxycholic acid Cholic acid (CDCA) (CA)
Peroxisomal functions • Bile acid biosynthesis • Fatty acid α - and β -oxidation • Plasmalogen biosynthesis • Glyoxylate metabolism • L-pipecolic acid oxidation
Fatty acid oxidation in peroxisomes • Very long-chain fatty acids ( ≥ C24:0) OH O • Branched-chain fatty acids (phytanic acid, pristanic acid) OH OH O • Bile acid intermediates (DHCA, THCA); Biosynthesis bile acids OH H O • Long-chain dicarboxylic acids • Polyunsaturated fatty acids; Biosynthesis C22:6 ω 3 and C22:5 ω 6
Fatty acid oxidation in peroxisomes β -oxidation
Peroxisomal disorders associated with bile acid abnormalities
Peroxisomal disorders associated with bile acid abnormalities • Peroxisome deficiency disorders (Zellweger spectrum) • α -methylacyl-CoA racemase deficiency (AMACR) • D-bifunctional protein deficiency (DBP) • Sterol carrier protein X (SCPx)
Peroxisome deficiency disorders Zellweger syndrome spectrum: • Zellweger syndrome • Neonatal adrenoleukodystrophy • Infantile Refsum disease Characterized by the absence of functional peroxisomes. Deficiency of multiple peroxisomal metabolic pathways. Caused by mutations in one of the 12 human PEX genes involved in peroxisome assembly.
Peroxisome deficiency disorders Clinical presentation: • Neonatal hypotonia • Seizures • Impaired vision, impaired hearing • Psycho-motor retardation • Dysmorphic features • Steatorrhea, fat-soluble vitamin deficiency • Liver disease
Peroxisome deficiency disorders Clinical presentation: • Liver disease • Hepatomegaly • Increased serum liver enzymes • Cholestasis • Proliferation of bile ducts • Fibrosis (eventually cirrhosis) • Steatosis • Hemosiderosis • Inflammation • Lesions of cholangioles
Peroxisome deficiency disorders � ↓ Plasmalogens � ↑ VLCFAs � ↑ Pristanic acid and phytanic acid (diet and age dependent) � ↑ DHCA , THCA, C29-dicarboxylic acid - Sum of DHCA, THCA, C29-dicarboxylic acid as a % of total bile acids ranges from 5-90%. - Pool size CDCA and CA is markedly reduced. - C27-bile acid intermediates only partly conjugated, whereas C24-bile acids normally conjugated. - C29-dicarboxylic acid poorly excreted in urine and bile.
Peroxisome deficiency disorders • Urine � Taurine conjugate of OH-THCA and di-OH-THCA ( m / z 572 and 588) � 5 β -cholestanepentol and 27-nor- 5 β -cholestanepentol glucuronide ( m / z 627 and 613) ↓
α -methylacyl-CoA racemase (AMACR) deficiency
α -methylacyl-CoA racemase (AMACR) deficiency Two clinical presentations: • The bile acid biosynthesis defect causes symptoms in childhood. • The accumulation of branched-chain fatty acids caused symptoms in adulthood.
α -methylacyl-CoA racemase (AMACR) deficiency Clinical presentation in children: • Fat-soluble vitamin deficiency (K, E, D) • Cholestatic liver disease
α -methylacyl-CoA racemase (AMACR) deficiency Clinical presentation in adult patients: • Adult-onset sensory motor neuropathy • Encephalopathy • Seizures • Postural and intention tremor • Retinitis pigmentosa, Cataracts
α -methylacyl-CoA racemase (AMACR) deficiency • Plasma � ↑ Pristanic acid and phytanic acid (diet and age dependent) � ↑ (25 R )-DHCA and (25 R )-THCA S R S R PBD AMACR
D-bifunctional protein (DBP) deficiency
D-bifunctional protein (DBP) deficiency Clinical presentation: • Neonatal hypotonia • Seizures • Impaired vision, impaired hearing • Psycho-motor retardation • Liver disease • Hepatomegaly • Cholestasis • Proliferation of bile ducts • Fibrosis • Steatosis • Hemosiderosis
D-bifunctional protein (DBP) deficiency • Plasma � ↑ VLCFAs � ↑ Pristanic acid and phytanic acid (diet and age dependent) � ↑ DHCA and THCA (in most but not all patients)
D-bifunctional protein (DBP) deficiency Three subgroups of DBP deficiency: • Type I: Combined DBP hydratase+dehydrogenase deficiency • Type II: Isolated DBP enoyl-CoA hydratase deficiency • Type III: Isolated DBP 3-hydroxyacyl-CoA dehydrogenase deficiency
Bile acid abnormalities and DBP deficiency Type I + II: DHCA, THCA, 24-ene-THCA, (24 S ,25 S )-24-OH-THCA Type III: (24 R ,25 R )-24-OH-THCA, (24 R ,25 S )-24-OH-THCA, (24 S ,25 S )-24-OH-THCA, DHCA, THCA, 24-ene-THCA
D-bifunctional protein (DBP) deficiency Patients with a longer survival tend to accumulate less DHCA and THCA, suggesting that residual DBP activity contributes to the extent of bile acid abnormalities in patients.
D-bifunctional protein (DBP) deficiency • Urine � Taurine conjugate of OH-24-ene-THCA and di-OH-24-ene- THCA (m/z 570 and 586)
Sterol carrier protein X (SCPx) deficiency
Sterol carrier protein X (SCPx) deficiency Clinical presentation: • Spasmodic torticollis with dystonic head tremor • Hyposmia, hypoacusis, nystagmus • No abnormalities upon ophthalmologic investigations • Sensory motor neuropathy • Slight cerebellar ataxia
Sterol carrier protein X (SCPx) deficiency • Plasma � ↑ Pristanic acid and phytanic acid � ↑ DHCA and THCA • Urine � Excretion of abnormal bile alcohol glucuronides
Sterol carrier protein X (SCPx) deficiency Glucuronide conjugates: •m/z 611 cholestanetetrol, of pentahydroxy-27nor-cholestan- 24one •m/z 613 27-nor-cholestanepentol •m/z 627 cholestanepentol, of hexahydroxy-27nor-cholestan- 24one •m/z 629 27-nor-cholestanehexol
Conclusion Bile acid abnormalities in peroxisomal disorders: • C24-bile acids are reduced → reduced bile flow → cholestasis • Accumulation of C27-bile acid intermediates → more hydrophobic, especially toxic, poorly conjugated, poorly excreted → cholestasis + liver injury • Bile acid therapy successful in relatively mild patients
Acknowledgments Thanks to many people at the laboratory Genetic Metabolic Diseases, AMC. Ronald J.A. Wanders Ries Duran Simone Denis Henk Overmars Albert Bootsma Henny Rusch Emma’s Children Hospital, AMC Bwee-Tien Poll The Peter Barth
Recommend
More recommend
Unleash a World of Digital Possibilities—Browse, Share, and Explore Content Without Boundaries