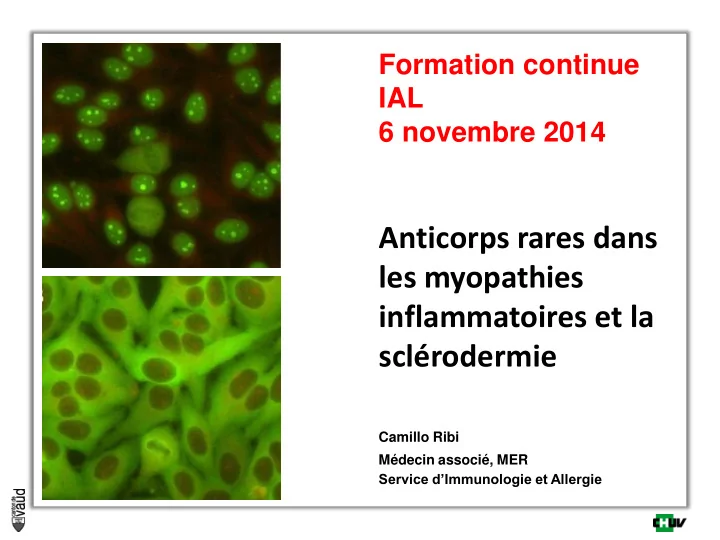
Formation continue IAL 6 novembre 2014 Anticorps rares dans les myopathies inflammatoires et la sclérodermie Camillo Ribi Médecin associé, MER Service d’Immunologie et Allergie
Dépistage auto-anticorps connectivites (LIA) Recherche d’ANA (=FAN) (Immunofluorescence indirecte sur lignée cellulaire Hep-2) Titre 1/80 Titre 1/320 Titre 1/80 – 1/160 Négatif homogène moucheté Ac anti-nucléosomes (ELISA) Ac. anti-nucléoprotéines (ECL) Négatif Positif Négatif Positif Spécificités (ECL) Ac anti-dsDNA SSA, SSB, RNP, Sm, Scl-70, Jo1
Dépistage auto-anticorps connectivites (LIA) fluorescence Recherche d’ANA (=FAN) cytoplasmique (Immunofluorescence indirecte sur lignée cellulaire Hep-2) Titre 1/80 Titre 1/320 Titre 1/80 – 1/160 centromère Négatif homogène moucheté nucléolaire Ac anti-nucléosomes (ELISA) Ac. anti-nucléoprotéines (ECL) dot sclerodermie Négatif Positif Négatif Positif Spécificités (ECL) dot myosites Ac anti-dsDNA SSA, SSB, RNP, Sm, Scl-70, Jo1
Immuno-dot / line blot disponibles au LIA Sclérose systémique Myopathies inflammatoires EUROLINE Systemic Sclerosis Profile Sclero-Poly-Synthétase Profile 12 dot
Types of autoimmune inflammatory myopathies PM IBM DM CTD anti- synthetases Immune-mediated necrotizing myositis
Features of immune-mediated myopathies adapté de Luo YB, Mastaglia FL. Biochim Biophys Acta 2014
Pathomechanisms in dermatomyositis Ischemic myopathy - deposition of immunoglobulins on intramuscular capillaries (?)* - complement (C5b-9) mediated injury to endothelial cells - depletion of the muscle capillary bed - muscle fibre necrosis - characteristic perifascicular pattern of muscle fibre atrophy * unclear how the complement pathway is activated in DM auto-antibodies targeting endothelial cell antigens have never been identified Inflammatory cells in perimysial and perivascular infiltrates: - B cells (upregulation of BAFF) - CD4 + T helper cells (Th1 and Th17) recruitement of mononuclear cells upregulation of MHC class I - interferon- α (IFNα ) producing plasmacytoid dendritic cells
Histological pattern of dermatomyositis A – C: haematoxylin and eosin D: MHC-I immunohistochemistry A. endomysial and perivascular mononuclear inflammatory infiltrate B. fragmentation of perimysial connective tissue and infiltration with granular mononuclear cells and macrophages C&D: perifascicular fibre regeneration and atrophy
Antibodies in inflammatory myopathies Various autoantibodies with high specificity Role in the pathogenesis of the muscle damage remains unclear Considerable racial and geographic differences in frequency Known to be influenced by genetic factors, particularly the HLA genotype Distinction of Myositis- specific Ab 30-58% of cases, mutually exclusive DM anti-Mi2, anti-TIF, anti-MDA5, anti-NXP2 Anti-synthétases anti-Jo1, anti-PL7, anti-PL12, anti-EJ Myosites nécrosantes anti-SRP, anti-HMGCR Myositis- associated Ab also found in other connective tissue diseases anti-SSA, anti-SSB, anti-U1RNP, anti-Ku, anti-PmScl...
Classic auto-antibodies in DM: Anti-Mi2 Target component of nucleosome remodelling deacetylase complex (NuRD) High levels of expression of Mi-2 in regenerating muscle cells and epidermis Some studies show correlation with level of UV light exposure -> Role in sustaining the auto-immune process ? Mainly in adult DM, rare in juvenile Reported frequency varies greatly (3 – 60%) Associated with: - Typical DM skin changes - Higher serum CK levels - Good response to treatment - Less likely to develop interstitial lung disease - Less likely to develop cancer
Newer antibodies in DM: TIF-1 γ and TIF- 1α Antibodies to transcription intermediary factor 1 γ and α (TIF - 1 γ, α) Target are 155/140 kDa nuclear proteins Indirect immunofluorescence: nuclear pattern Pathophysiological role of anti-TIF Ab not yet understood TIF generated as part of an anti-tumoral response ? Antibodies may cross-react with muscle/skin expressing high levels of TIF anti- TIF γ mostly coexist with anti - TIFα Both in juvenile and adult DM Positivity rate of 16 – 23% Adult patients have a higher incidence of malignancy
Newer antibodies in DM: anti-MDA5 (CADM-140) Targets cytoplasmic RNA-specific helicase belonging to RIG-I-like receptor family Now known as ‘melanoma differentiation - associated gene 5’ (MDA5) MDA5 participates in the innate immune response to viruses Potential activator of type I IFN transcription First described in Japanese patients Considerable racial and ethnic variation in frequency and associated phenotype About 7% of DM (USA) usually: more severe skin manifestations (skin and oral ulcerations) higher incidence of rapidly progressive interstitial lung disease less muscle involvement (lower serum CK levels) picture sometimes resembling the anti-synthetase syndrome
MDA5: a cytosolic member of PRR Bowie AG. Nature Reviews Immunology 2008
Clinical features of MDA5 + dermatomyositis Fiorentino D. J Am Acad Dermatol 2011
Typical skin manifestations of anti-MDA5+ DM Fiorentino D. J Am Acad Dermatol 2011
Newer antibodies in DM: Anti-NXP-2 (MJ, p-180) Target: nuclear matrix protein 2 (NXP-2) Indirect immunofluorescence: nuclear Present in up to 25% of cases of juvenile DM Ethnic/geographic variability in frequency (Italy: 30% adult DM, Japan <2%) More common in patients of younger age Associated with More severe muscle weakness Calcinosis Joint contractures Intestinal vasculitis In adults: increased incidence of neoplasia, particularly in males
« Polymyositis »
« Polymyositis » Most of the time part of a systemic autoimmune disease Mainly in adults, very rare in childhood Painless proximal myopathy with subacute onset Subvariants: Distal weakness mainly in the upper limbs, with good response to treatment Involvement of the paraspinal muscles with head drop (camptocormia) Usually responsive to corticosteroids and immunosuppression Pathogenesis: CD8 + T cell predominant lymphocytes invading non-necrotic fibres Interaction with antigen-presenting MHC-I molecules Doubt as to whether there is a common self-antigen Cytotoxicity trough release of perforin
Histological pattern of overlap-myositis A ,C: haematoxylin & eosin B: MHC-I immunohistochemistry D: modified Gomori trichrome. A – D: endomysial mononuclear inflammatory infiltrate myofibre invasion by mononuclear cells (C, arrowhead) myofibre necrosis (C, D); and diffuse MHC-I antigen expression (B).
Autoantibodies in PM Anti-synthetase syndrome n.b. anti-Jo1 reported in up to 16% of DM Antibodies to synthetase are considered myositis-specific Mixed connective tissue disease (MCTD) high titres of antibodies to 70-kD ribonuclear protein complex (anti-U1 snRNP) features of systemic sclerosis / SLE such as Raynaud's phenomenon Sclerodactyly Synovitis Scleroderma – myositis overlap syndrome only a small minority of cases of scleroderma presence of antibodies to the nucleolar PM-Scl antigen complex (75 or 100kD)
Anti-synthetase syndrome Characterized by antibodies to one of the tRNA synthetases (cytoplasma) Anti-Jo1 (anti-histidyl tRNA synthetase) most prevalent (20% of cases) Anti-Jo1 much more (5x) frequent than all the other anti-synthetase Ab Histopathologically: Perimysial connective tissue damage CD68 + histiocytes and perivascular CD4 + T cells perifascicular fibre necrosis and regeneration Clinically myositis + other systemic features, including - interstitial lung disease (sometimes precedes myositis !) - Raynaud's phenomenon - deforming arthropathy - skin changes in the hands Condition is relatively resistant to treatment
Immune-mediated necrotizing myopathy Heterogeneous group of acquired myopathies Distinguishable from DM and PM on clinical and pathological grounds Histopathologically: Myofibre necrosis Inflammation is usually lacking Immunostaining for C5b-9 and MHC-I in some cases Amenable to treatment with immune therapies May be classified into different categories, associated with - malignancy - connective tissue diseases - viral infections (HIV, HCV) - specific auto-antibodies: anti-SRP, anti-HMGCR and anti-synthetase These antibodies are putatively involved in the pathogenesis...
Immune-mediated necrotizing myopathy A, C: haemotoxylin & eosin B: MHC-I immunohistochemistry D: CD163 immunohistochemistry A,B: Associated with prolonged statin therapy; C,D: idiopathic case A: muscle fibre necrosis with sparse inflammatory infiltrate and B: diffuse MHC-I expression C: necrotising myopathy with D: scattered macrophagic infiltrate.
Myopathy associated with anti-SRP antibody Anti-SRP previously considered a specific antibodies associated with PM or DM Myopathy associated with anti-SRP antibody now stands as a separate entity Infrequently associated with other autoimmune diseases Clinically: rapidly progressive > chronic proximal muscle weakness high serum CK levels relative resistance to treatment with conventional immune therapies Histopathologically: - active necrotising myopathy - inconspicuous or absent inflammatory infiltrate and MCH-I up-regulation - occasionnally C5b-9 deposition on endomysial capillaries and muscle fibres - predominance of macrophages
Recommend
More recommend
Unleash a World of Digital Possibilities—Browse, Share, and Explore Content Without Boundaries